

Anti-Prion Systems in Saccharomyces cerevisiae Turn an Avalanche of Prions into a Flurry
Abstract
1. What Are Prions?
2. What Do Prions Do in the Host?
3. How Does the Host Cell Deal with Prions?
3.1. Btn2p and Cur1p Act on the [URE3] Prion
3.2. Hsp104 at the Normal Level Acting on the [PSI+] Prion
3.3. Inositol Polyphosphates Acting on [PSI+] Prion Propagation
3.4. Nonsense-Mediated mRNA Decay Proteins Acting on [PSI+]
3.5. Ribosome-Associated Chaperones Acting on the [PSI+] Prion
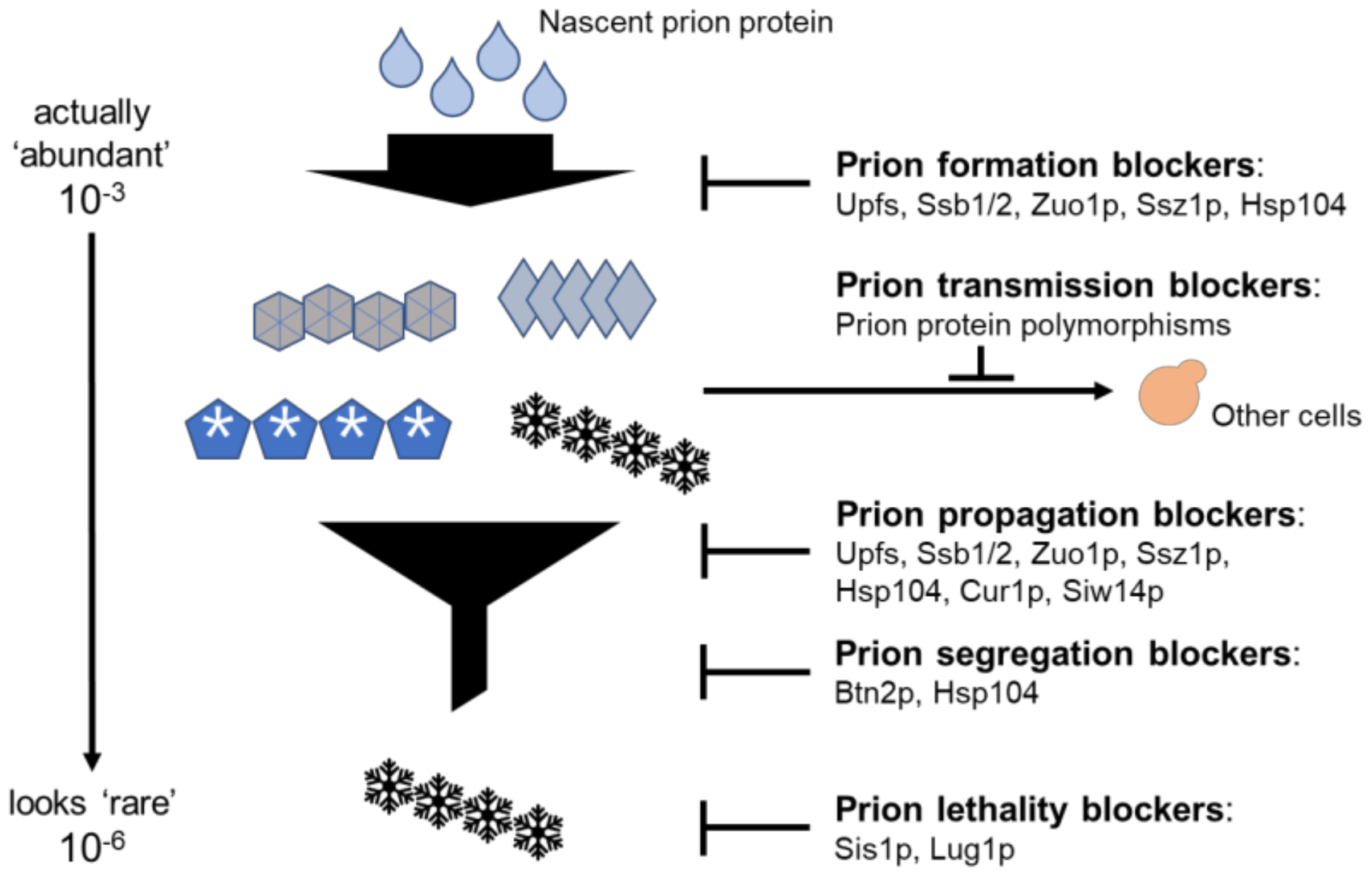
3.6. Anti-Prion Systems Turn an Avalanche of Prions into a Flurry
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in S. cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.R.; Kowal, A.S.; Shirmer, E.C.; Patino, M.M.; Liu, J.-J.; Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997, 89, 811–819. [Google Scholar] [CrossRef]
- Edskes, H.K.; Gray, V.T.; Wickner, R.B. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc. Natl. Acad. Sci. USA 1999, 96, 1498–1503. [Google Scholar] [CrossRef]
- Roberts, B.T.; Wickner, R.B. A class of prions that propagate via covalent auto-activation. Genes Dev. 2003, 17, 2083–2087. [Google Scholar] [CrossRef]
- King, C.-Y.; Diaz-Avalos, R. Protein-only transmission of three yeast prion strains. Nature 2004, 428, 319–323. [Google Scholar] [CrossRef]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Smejkal, T.; Itakura, A.K.; Garcia, D.M.; Jarosz, D.F. A non-amyloid prion particle that activates a heritable gene expression program. Mol. Cell 2020, 77, 251–265. [Google Scholar] [CrossRef]
- Cox, B.S. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965, 20, 505–521. [Google Scholar] [CrossRef]
- Alper, T.; Haig, D.A.; Clarke, M.C. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966, 22, 278–284. [Google Scholar] [CrossRef]
- Lacroute, F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 1971, 106, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Masison, D.C.; Wickner, R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.; Kryndushkin, D.; Wickner, R.B. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. USA 2011, 108, 5337–5341. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.B. Scrapie Disease in Sheep–Historical, Clinical, Epidemiological, Pathological and Practical Aspects of the Natural Disease; Academic Press: London, UK, 1983; p. 192. [Google Scholar]
- Wickner, R.B. Scrapie in ancient China? Science 2005, 309, 874. [Google Scholar] [CrossRef]
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef]
- Diringer, H.; Gelderblom, H.; Hilmert, H.; Ozel, M.; Edelbluth, C.; Kimberlin, R.H. Scrapie infectivity, fibrils and low molecular weight protein. Nature 1983, 306, 476–478. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-b pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for a-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal propagation of pathologic a-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Ter-Avanesyan, M.D.; Dagkesamanskaya, A.R.; Kushnirov, V.V.; Smirnov, V.N. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 1994, 137, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Son, M.; Edskes, B.K. Prion variants of yeast are numerous, mutable, and segregate on growth, affecting prion pathogenesis, transmission barriers and sensitivity to anti-prioin systems. Viruses 2019, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Wu, S.; Niznikiewicz, M. Innate immunity to prions: Anti-prion systems turn a tsunami of prions into a slow drip. Curr. Genet. 2021, 67, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Lund, P.M.; Cox, B.S. Reversion analysis of [psi-] mutations in Saccharomyces cerevisiae. Genet. Res. 1981, 37, 173–182. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Prions, chaperones, and proteastasis in yeast. Cold Spring Harb. Perspect. Biol. 2017, 1, a023663. [Google Scholar] [CrossRef]
- Masison, D.C.; Reidy, M. Yeast prions are useful for studying protein chaperones and protein quality control. Prion 2015, 9, 174–183. [Google Scholar] [CrossRef]
- Ross, E.D.; Edskes, H.K.; Terry, M.J.; Wickner, R.B. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. USA 2005, 102, 12825–12830. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.D.; Minton, A.P.; Wickner, R.B. Prion domains: Sequences, structures and interactions. Nat. Cell Biol. 2005, 7, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Shewmaker, F.; Bateman, D.A.; Edskes, H.E.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E. Yeast prions: Structure, biology and prion-handling systems. Microbiol. Mol. Biol. Rev. 2015, 79, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.E.; Bateman, D.A.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E.; Mukhamedova, M. Yeast Prions: Proteins templating conformation and an anti-prion system. PLoS Pathog. 2015, 11, e1004584. [Google Scholar] [CrossRef]
- DePace, A.H.; Santoso, A.; Hillner, P.; Weissman, J.S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998, 93, 1241–1252. [Google Scholar] [CrossRef]
- Liu, J.J.; Lindquist, S. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 1999, 400, 573–576. [Google Scholar] [CrossRef]
- Chen, B.; Bruce, K.L.; Newnam, G.P.; Gyoneva, S.; Romanyuk, A.V.; Chernoff, Y.O. Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol. Microbiol. 2010, 76, 1483–1499. [Google Scholar] [CrossRef]
- Diaz-Avalos, R.; King, C.Y.; Wall, J.S.; Simon, M.; Caspar, D.L.D. Strain-specific morphologies of yeast prion amyloids. Proc. Natl Acad. Sci. USA 2005, 102, 10165–10170. [Google Scholar] [CrossRef]
- Shewmaker, F.; Wickner, R.B.; Tycko, R. Amyloid of the prion domain of Sup35p has an in-register parallel b-sheet structure. Proc. Natl. Acad. Sci. USA 2006, 103, 19754–19759. [Google Scholar] [CrossRef]
- Baxa, U.; Wickner, R.B.; Steven, A.C.; Anderson, D.; Marekov, L.; Yau, W.-M.; Tycko, R. Characterization of b-sheet structure in Ure2p1-89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry 2007, 46, 13149–13162. [Google Scholar] [CrossRef]
- Wickner, R.B.; Dyda, F.; Tycko, R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register b-sheet structure. Proc. Natl. Acad. Sci. USA 2008, 105, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Thurber, K.R.; Shewmaker, F.; Wickner, R.B.; Tycko, R. Measurement of amyloid fibril mass-per-length by tilted-beam transmission electron microscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 14339–14344. [Google Scholar] [CrossRef] [PubMed]
- Gorkovskiy, A.; Thurber, K.R.; Tycko, R.; Wickner, R.B. Locating folds of the in-register parallel b-sheet of the Sup35p prion domain infectious amyloid. Proc. Natl. Acad. Sci. USA 2014, 111, E4615–E4622. [Google Scholar] [CrossRef]
- Ohhashi, Y.; Yamaguchi, Y.; Kurahashi, H.; Kamatari, Y.O.; Sugiyama, S.; Uluca, B.; Piechatzek, T.; Komi, Y.; Shida, T.; Muller, H.; et al. Molecular basis for diversification of yeast prion strain conformation. Proc. Natl. Acad. Sci. USA 2018, 115, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.-H.; King, C.-Y. Amino acid proximities in two Sup35 prion strains revealed by chemical cross-linking. J. Biol. Chem. 2015, 290, 25062–25071. [Google Scholar] [CrossRef]
- Wang, J.; Park, G.; Lee, Y.K.; Nguyen, M.; San Fung, T.; Lin, T.Y.; Hsu, F.; Guo, Z. Spin label scanning reveals likely locations of β-strands in the amyloid fibrils of the Ure2 prion domain. ACS Omega 2020, 5, 5984–5993. [Google Scholar] [CrossRef]
- Dergalev, A.A.; Alexandrov, A.I.; Ivannikov, R.I.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast Sup35 prion structure: Two types, four parts, many variants. Int. J. Mol. Sci. 2019, 20, 2633. [Google Scholar] [CrossRef]
- Wickner, R.B.; Edskes, H.K.; Shewmaker, F.; Nakayashiki, T. Prions of fungi: Inherited structures and biological roles. Nat. Rev. Microbiol. 2007, 5, 611–618. [Google Scholar] [CrossRef]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Shewmaker, F.; Wickner, R.B. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008, 27, 2725–2735. [Google Scholar] [CrossRef]
- Wickner, R.B.; Beszonov, E.; Bateman, D.A. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc. Natl. Acad. Sci. USA 2014, 111, E2711–E2720. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.-I.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 at normal levels cures many [PSI+] variants in a process promoted by Sti1p, Hsp90 and Sis1p. Proc. Natl. Acad. Sci. USA 2017, 114, E4193–E4202. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Kelly, A.C.; Bezsonov, E.E.; Edskes, H.E. Prion propagation is controlled by inositol polyphosphates. Proc. Natl. Acad. Sci. USA 2017, 114, E8402–E8410. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Wickner, R.B. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2018, 115, E1184–E1193. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Wickner, R.B. Normal levels of ribosome-associated chaperones cure two groups of [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2020, 117, 26298–26306. [Google Scholar] [CrossRef] [PubMed]
- Bateman, D.A.; Wickner, R.B. [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: Intraspecies ’species barriers’. Genetics 2012, 190, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, P.A.; Reidy, M.; Masison, D.C. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 2011, 188, 565–577. [Google Scholar] [CrossRef]
- Kumar, J.; Reidy, M.; Masison, D.C. Yeast J-protein Sis1 prevents prion toxicity by moderating depletion of prion protein. Genetics 2021, 219, iyab129. [Google Scholar] [CrossRef]
- Edskes, H.E.; Mukhamedova, M.; Edskes, B.K.; Wickner, R.B. Hermes transposon mutagenesis shows [URE3] prion pathology prevented by a ubiquitin-targeting protein: Evidence for carbon/nitrogen assimilation cross-talk and a second function for Ure2p. Genetics 2018, 209, 789–800. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Ihrke, G.; Piermartiri, T.C.; Shewmaker, F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 2012, 86, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Malinovska, L.; Kroschwald, S.; Munder, M.C.; Richter, D.; Alberti, S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spaciotemporal distribution of protein aggregates. Mol. Biol. Cell 2012, 23, 3041–3056. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.B.; Ho, C.T.; Winkler, J.; Khokhrina, M.; Neuner, A.; Mohamed, M.Y.; Guilbride, D.L.; Richter, K.; Lisby, M.; Scheibel, E.; et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 2015, 34, 778–797. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82. [Google Scholar] [CrossRef]
- Tessarz, P.; Mogk, A.; Bukau, B. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol. Microbiol. 2008, 68, 87–97. [Google Scholar] [CrossRef]
- Hung, G.C.; Masison, D.C. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 2006, 173, 611–620. [Google Scholar] [CrossRef]
- Gerstmann, J. Uber ein noch nicht beschriebenes Reglexphanomen bei einer Erkrankung des zerebellum systems. Wien Med. Wehnschr. 1928, 78, 906–908. [Google Scholar]
- Lum, R.; Tkach, J.M.; Vierling, E.; Glover, J.R. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem. 2004, 279, 29139–29146. [Google Scholar] [CrossRef]
- Lum, R.; Niggemann, M.; Glover, J.R. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J. Biol. Chem. 2008, 283, 30139–30146. [Google Scholar] [CrossRef]
- Park, Y.N.; Zhou, X.; Yim, Y.I.; Todor, H.; Ellerbrock, R.; Reidy, M.; Eisenberg, E.; Masison, D.C.; Greene, L.E. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot. Cell 2014, 13, 635–647. [Google Scholar] [CrossRef]
- Ness, F.; Cox, B.; Wonwigkam, J.; Naeimi, W.R.; Tuite, M.F. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI+] propagons. Mol. Microbiol. 2017, 104, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Helsen, C.W.; Glover, J.R. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J. Biol. Chem. 2012, 287, 542–556. [Google Scholar] [CrossRef]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Steidle, E.A.; Chong, L.S.; Wu, M.; Crooke, E.; Fiedler, D.; Resnick, A.C.; Rolfes, R.J. A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 protein selectively cleaves the b-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5). J. Biol. Chem. 2016, 291, 6772–6783. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Jacobson, A. Nonsense-mediated mRNA decay: Degradation of defective transcripts is only part of the story. Ann. Rev. Genet. 2015, 49, 339–366. [Google Scholar] [CrossRef]
- Czaplinski, K.; Ruiz-Echevarria, M.J.; Paushkin, S.V.; Han, X.; Weng, Y.; Perlick, H.A.; Dietz, H.C.; Ter-Avanesyan, M.D.; Peltz, S.W. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 1998, 12, 1665–1677. [Google Scholar] [CrossRef]
- Wang, W.; Czaplinski, K.; Rao, Y.; Peltz, S.W. The role of Upf proteins in modulating the translation read-through of nonsense-containing transcripts. EMBO J. 2001, 20, 880–890. [Google Scholar] [CrossRef]
- He, F.; Brown, A.H.; Jacobson, A. Upf1p, Nmd2p and Upf3p are interacting components of the yeast nonsense-mediated mRNA decay pathway. Mol. Cell. Biol. 1997, 17, 1580–1594. [Google Scholar] [CrossRef]
- Nelson, R.J.; Ziegilhoffer, T.; Nicolet, C.; Werner-Washburne, M.; Craig, E.A. The translation machinery and 70 kDal heat shock protein cooperate in protein synthesis. Cell 1992, 71, 97–105. [Google Scholar] [CrossRef]
- Pfund, C.; Lopez-Hoyo, N.; Ziegelhoffer, T.; Schilke, B.A.; Lopez-Buesa, P.; Walter, W.A.; Wiedmann, M.; Craig, E.A. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 1998, 17, 3981–3989. [Google Scholar] [CrossRef]
- Gautschi, M.; Lilie, H.; Fünfschilling, U.; Mun, A.; Ross, S.; Lithgow, T.; Rücknagel, P.; Rospert, S. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc. Natl. Acad. Sci. USA 2001, 98, 3762–3767. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, M.; Mun, A.; Ross, S.; Rospert, S. A functional chaperone triad on the yeast ribosome. Proc. Natl. Acad. Sci. USA 2002, 99, 4209–4214. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Newnam, G.P.; Kumar, J.; Allen, K.; Zink, A.D. Evidence for a protein mutator in yeast: Role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI+] prion. Mol. Cell. Biol. 1999, 19, 8103–8112. [Google Scholar] [CrossRef] [PubMed]
- Kiktev, D.A.; Melomed, M.M.; Lu, C.D.; Newnam, G.P.; Chernoff, Y.O. Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol. Microbiol. 2015, 96, 621–632. [Google Scholar] [CrossRef]
- Amor, A.J.; Castanzo, D.T.; Delany, S.P.; Selechnik, D.M.; van Ooy, A.; Cameron, D.M. The ribosome-associated complex antagonizes prion formation in yeast. Prion 2015, 9, 144–164. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Kiktev, D.A. Dual role of ribosome-associated chaperones in prion formation and propagation. Curr. Genet. 2016, 62, 677–685. [Google Scholar] [CrossRef]
- Howie, R.L.; Jay-Garcia, L.M.; Kiktev, D.A.; Faber, Q.L.; Murphy, M.; Rees, K.A.; Sachwani, N.; Chernoff, Y.O. Role of the Cell Asymmetry Apparatus and Ribosome-Associated Chaperones in the Destabilization of a Saccharomyces cerevisiae Prion by Heat Shock. Genetics 2019, 212, 757–771. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.-M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.-L.; Serban, D.; Carlson, G.A.; et al. Transgenic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Galkin, A.P.; Lewitin, E.; Chernova, T.A.; Newnam, G.P.; Belenkiy, S.M. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol. Microbiol. 2000, 35, 865–876. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Kochneva-Pervukhova, N.V.; Cechenova, M.B.; Frolova, N.S.; Ter-Avanesyan, M.D. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000, 19, 324–331. [Google Scholar] [CrossRef]
- Santoso, A.; Chien, P.; Osherovich, L.Z.; Weissman, J.S. Molecular basis of a yeast prion species barrier. Cell 2000, 100, 277–288. [Google Scholar] [CrossRef]
- Edskes, H.K.; Wickner, R.B. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full-length protein. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S4), 16384–16391. [Google Scholar] [CrossRef] [PubMed]
- Luke, M.M.; Sutton, A.; Arndt, K.T. Characterization of SIS1, a Saccharomyces cerevisiae homolog of bacterial dnaJ proteins. J. Cell Biol. 1991, 114, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Arndt, K.T. The yeast SIS1 protein, a DnaJ homolog, is required for initiation of translation. Cell 1993, 73, 1175–1186. [Google Scholar] [CrossRef]
- Higurashi, T.; Hines, J.K.; Sahi, C.; Aron, R.; Craig, E.A. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. USA 2008, 105, 16596–16601. [Google Scholar] [CrossRef]
- Hua, Z.; Vierstra, R.D. The cullin-ring ubiquitin-protein ligases. Ann. Rev. Plant Path. 2011, 62, 299–334. [Google Scholar] [CrossRef]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef]
- Son, M.; Wickner, R.B. Antiprion systems in yeast cooperate to cure or prevent the generation of nearly all [PSI+] and [URE3] prions. Proc. Natl. Acad. Sci. USA 2022, 119, e2205500119. [Google Scholar] [CrossRef]
- Saupe, S.J. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Sem. Cell Dev. Biol. 2011, 22, 460–468. [Google Scholar] [CrossRef]
- Saupe, S.J.; Daskalov, A. The [Het-s] prion, an amyloid fold as a cell death activation trigger. PLoS Pathog. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Itakura, A.K.; Chakravarty, A.K.; Jakobson, C.M.; Jarosz, D.F. Widespread prion-based control of growth and differentiation strategies in Saccharomyces cerevisiae. Mol. Cell 2020, 77, 266–278.e6. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R.; Wickner, R.B. Molecular structures of amyloid and prion fibrils: Consensus vs. controversy. Acc. Chem. Res. 2013, 46, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 2014, 23, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes a-synuclein aggregation and motor impairment in mice. eLife 2020, 9, e53111. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Protein | Target Prion | Mechanism | Reference |
|---|---|---|---|
| Btn2p | [URE3] | Prion curing by sequestration of amyloid filaments | [51,52] |
| Cur1p | [URE3] | Prion curing with unknown mechanisms | [51,52] |
| Hsp104 | [PSI+] | Blocking generation and propagation | [53,54] |
| Siw14p | [PSI+] | Prion curing by the regulation of inositol poly/pyrophosphates | [55] |
| Upf1, 2, 3p | [PSI+] | Prion curing by complex formation with Sup35p | [56] |
| Ssb1/2p, Zuo1p, Ssz1p | [PSI+] | Prion curing by the protection of polypeptides from misfolding | [57] |
| Prion proteinpolymorphisms | [PSI+] | Intraspecies barrier to prion transmission by prion protein sequence differences | [58] |
| Sis1p | [PSI+] | Reducing a prion’s toxicity by helping Sup35 solubility | [59,60] |
| Lug1p | [URE3] | Reduction in a prion’s toxicity due to a functional defect of Ure2p in [URE3] cells. | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, M.; Wickner, R.B. Anti-Prion Systems in Saccharomyces cerevisiae Turn an Avalanche of Prions into a Flurry. Viruses 2022, 14, 1945. https://doi.org/10.3390/v14091945
Son M, Wickner RB. Anti-Prion Systems in Saccharomyces cerevisiae Turn an Avalanche of Prions into a Flurry. Viruses. 2022; 14(9):1945. https://doi.org/10.3390/v14091945
Chicago/Turabian StyleSon, Moonil, and Reed B. Wickner. 2022. "Anti-Prion Systems in Saccharomyces cerevisiae Turn an Avalanche of Prions into a Flurry" Viruses 14, no. 9: 1945. https://doi.org/10.3390/v14091945
APA StyleSon, M., & Wickner, R. B. (2022). Anti-Prion Systems in Saccharomyces cerevisiae Turn an Avalanche of Prions into a Flurry. Viruses, 14(9), 1945. https://doi.org/10.3390/v14091945