
The Second Human Pegivirus, a Non-Pathogenic RNA Virus with Low Prevalence and Minimal Genetic Diversity

 , , and
, , and
Abstract
1. Introduction
2. Prevalence of HPgV-2
3. Pathogenesis of HPgV-2
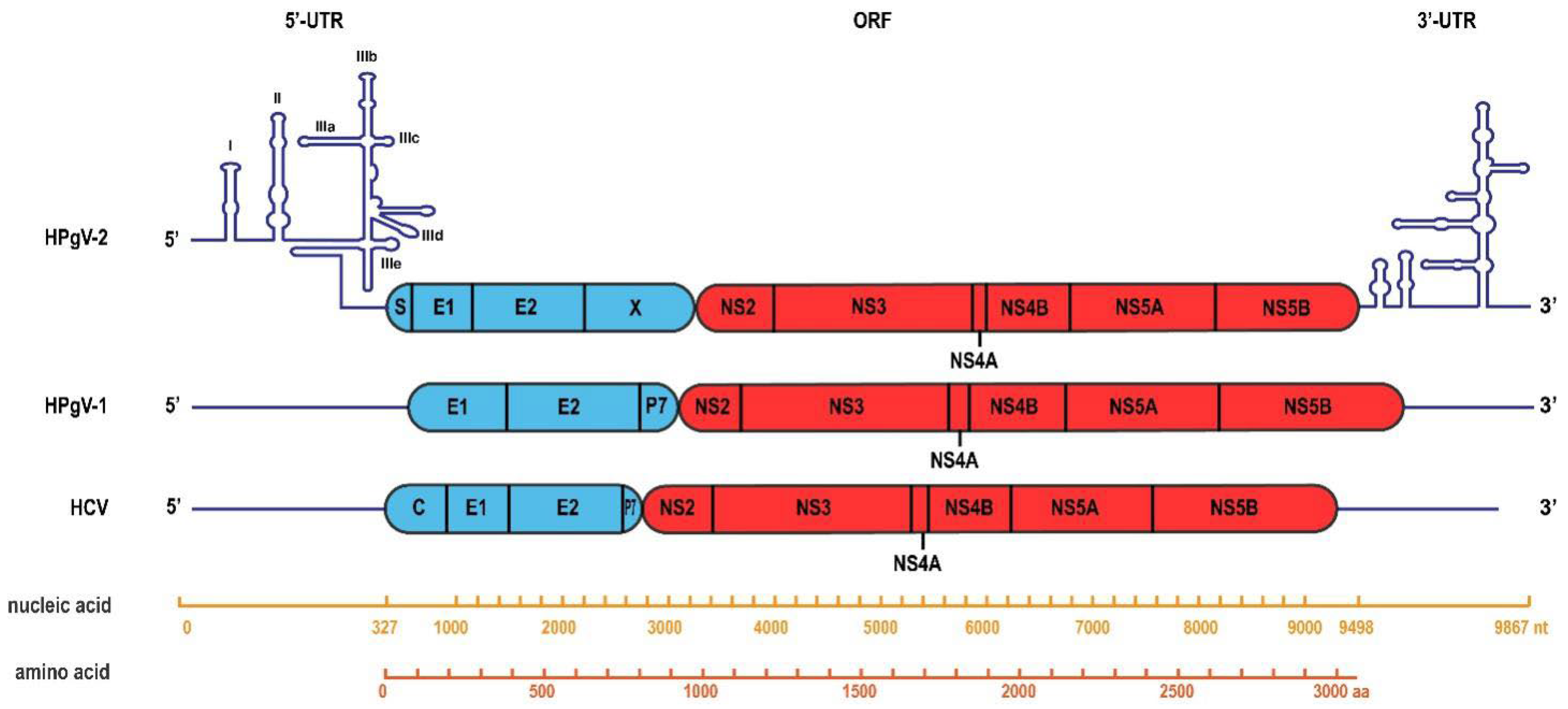
4. HPgV-2 Genome Organization
5. Minimum Genetic Diversity of HPgV-2
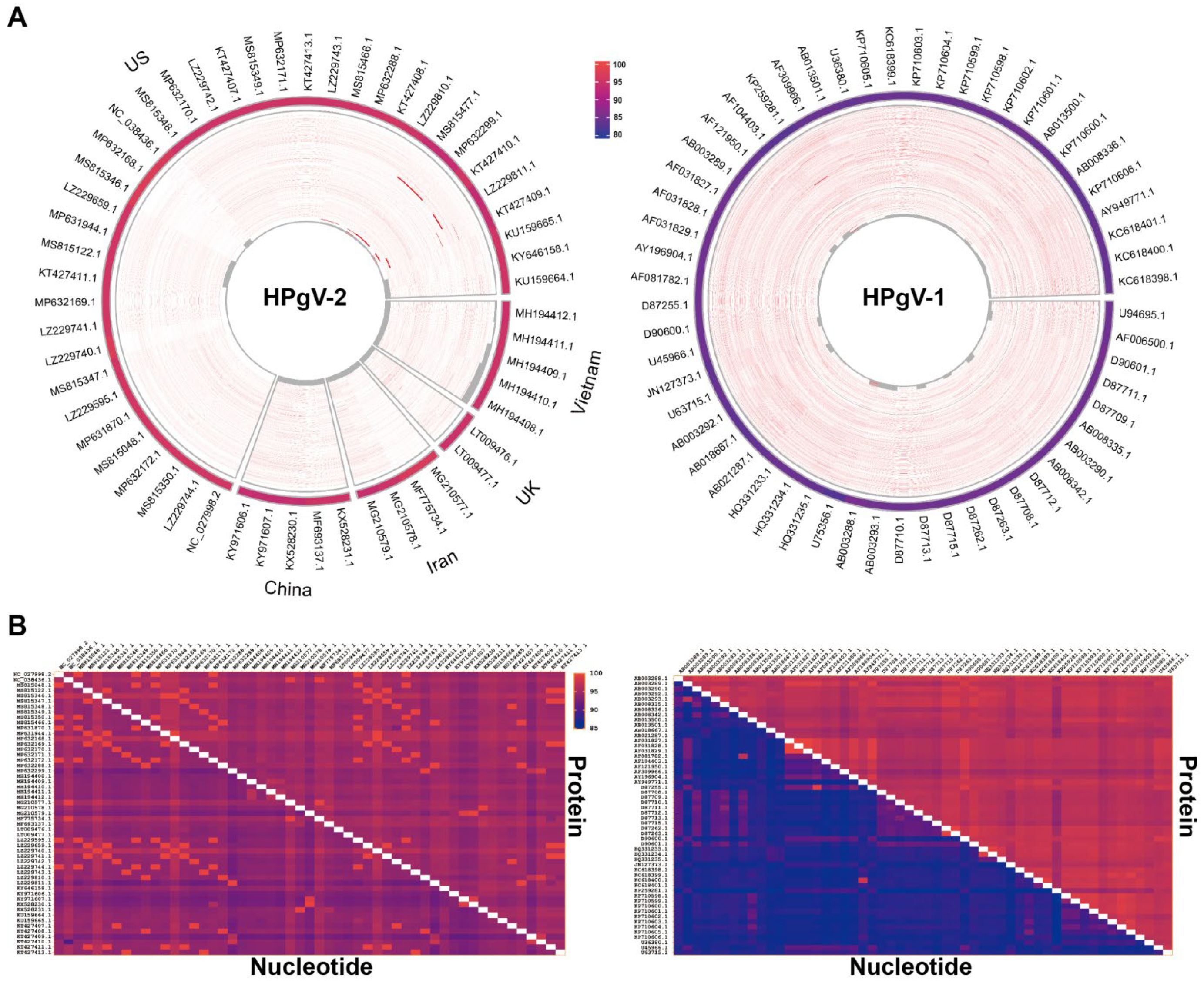
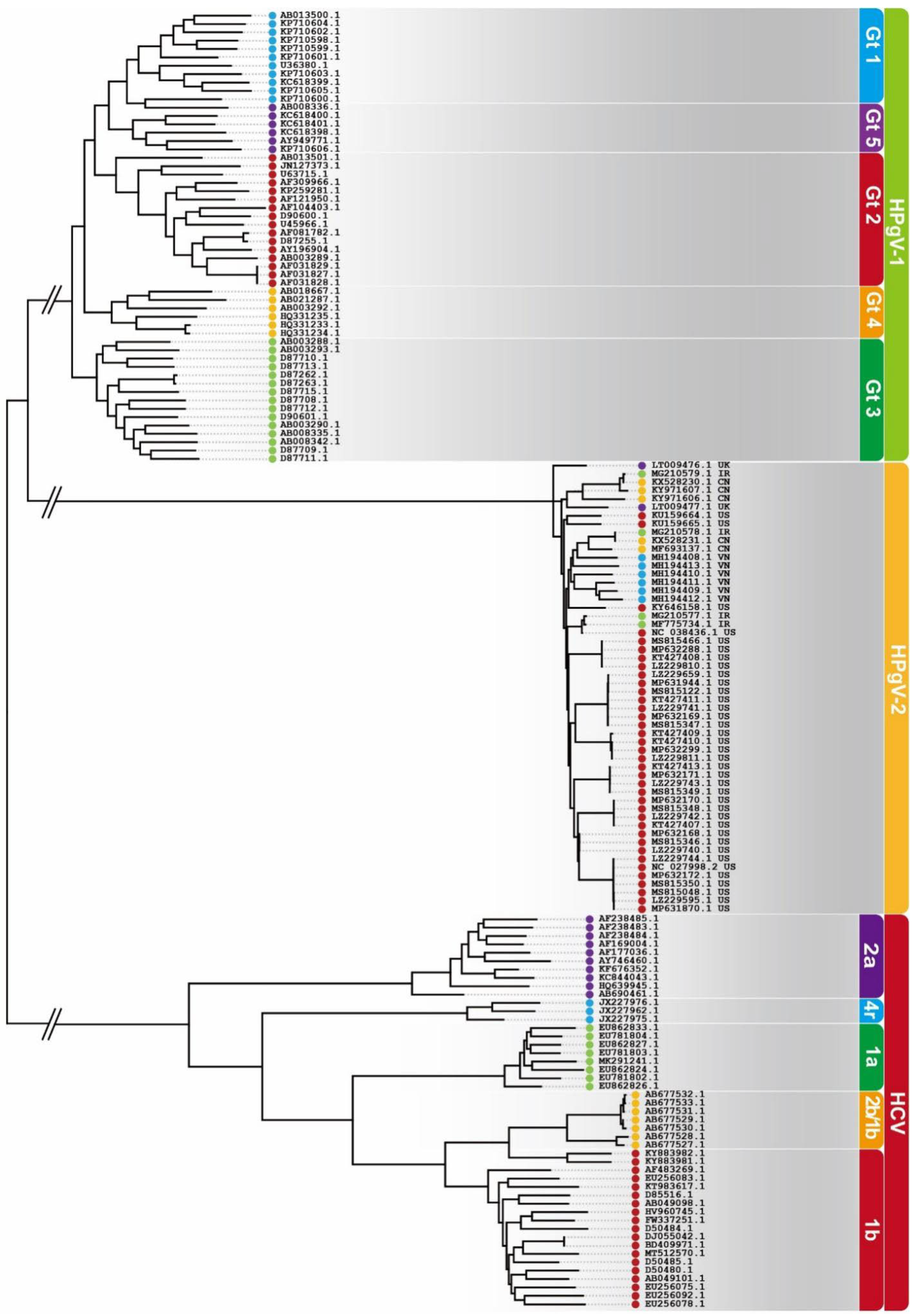
5.1. Conservation of HPgV-2 Genome
5.2. Stability of HPgV-2 Genome
5.3. HPgV-2 Shows Less Intra-Host Diversity than HCV
6. Possible Explanation for HPgV-2’s Genome Fidelity
7. Inspiration from HPgV-2
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simons, J.N.; Pilot-Matias, T.J.; Leary, T.P.; Dawson, G.J.; Desai, S.M.; Schlauder, G.G.; Muerhoff, A.S.; Erker, J.C.; Buijk, S.L.; Chalmers, M.L. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA 1995, 92, 3401–3405. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, R.A.; Dawson, G.J.; Knigge, M.F.; Melvin, S.L.; Heynen, C.A.; Kyrk, C.R.; Young, C.E.; Carrick, R.J.; Schlauder, G.G.; Surowy, T.K.; et al. Seroprevalence of GB virus C and persistence of RNA and antibody. J. Med. Virol. 1997, 53, 167–173. [Google Scholar] [CrossRef]
- Mohr, E.L.; Stapleton, J.T. GB virus type C interactions with HIV: The role of envelope glycoproteins. J. Viral Hepat. 2009, 16, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Stapleton, J.T. GB virus C: The good boy virus? Trends Microbiol. 2012, 20, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Schwarze-Zander, C.; Blackard, J.T.; Rockstroh, J.K. Role of GB virus C in modulating HIV disease. Expert Rev. Anti-Infect. Ther. 2012, 10, 563–572. [Google Scholar] [CrossRef]
- Tillmann, H.L.; Manns, M.P. GB virus-C infection in patients infected with the human immunodeficiency virus. Antivir. Res. 2001, 52, 83–90. [Google Scholar] [CrossRef]
- Lauck, M.; Bailey, A.L.; Andersen, K.G.; Goldberg, T.L.; Sabeti, P.C.; O’Connor, D.H. GB virus C coinfections in west African ebola patients. J. Virol. 2015, 89, 2425–2429. [Google Scholar] [CrossRef]
- Kapoor, A.; Kumar, A.; Simmonds, P.; Bhuva, N.; Chauhan, L.S.; Lee, B.; Sall, A.A.; Jin, Z.; Morse, S.S.; Shaz, B.; et al. Virome analysis of transfusion recipients reveals a novel human virus that shares genomic features with hepaciviruses and pegiviruses. mBio 2015, 6, e01466-15. [Google Scholar] [CrossRef]
- Berg, M.G.; Lee, D.; Coller, K.; Frankel, M.; Aronsohn, A.; Cheng, K.; Forberg, K.; Marcinkus, M.; Naccache, S.N.; Dawson, G.; et al. Discovery of a novel human pegivirus in blood associated with HEPATITIS C virus Co-infection. PLoS Pathog. 2015, 11, e1005325. [Google Scholar] [CrossRef]
- Bonsall, D.; Gregory, W.F.; Ip, C.L.; Donfield, S.; Iles, J.; Ansari, M.A.; Piazza, P.; Trebes, A.; Brown, A.; Frater, J.; et al. Evaluation of viremia frequencies of a novel human pegivirus by using bioinformatic screening and PCR. Emerg. Infect. Dis. 2016, 22, 671–678. [Google Scholar] [CrossRef]
- Wang, H.; Wan, Z.; Xu, R.; Guan, Y.; Zhu, N.; Li, J.; Xie, Z.; Lu, A.; Zhang, F.; Fu, Y.; et al. A novel human pegivirus, HPgV-2 (HHpgV-1), is tightly associated with hepatitis C virus (HCV) infection and HCV/human immunodeficiency virus type 1 coinfection. Clin. Infect. Dis. 2017, 66, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Skewes-Cox, P.; Zhong, W.; Ganem, D.E.; Divers, T.J.; Van Blaricum, A.J.; Tennant, B.C.; Kistler, A.L. Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. Proc. Natl. Acad. Sci. USA 2013, 110, E1407–E1415. [Google Scholar] [CrossRef]
- Kandathil, A.J.; Breitwieser, F.P.; Sachithanandham, J.; Robinson, M.; Mehta, S.H.; Timp, W.; Salzberg, S.L.; Thomas, D.L.; Balagopal, A. Presence of human Hepegivirus-1 in a cohort of people who inject drugs. Ann. Intern. Med. 2017, 167, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Anh, N.T.; Hong, N.T.T.; Nhu, L.N.T.; Thanh, T.T.; Anscombe, C.; Chau, L.N.; Lau, C.-Y.; Limmathurotsakul, D.; Chau, N.V.V.; Van Doorn, H.R.; et al. Detection and characterization of human pegivirus 2, vietnam. Emerg. Infect. Dis. 2018, 24, 2063–2067. [Google Scholar] [CrossRef] [PubMed]
- Forberg, K.; Rodgers, M.A.; Dawson, G.J.; Sauleda, S.; Olivo, A.; Vallari, A.; Bes, M.; Piron, M.; Cloherty, G.A.; Berg, M.G. Human pegivirus 2 exhibits minimal geographic and temporal genetic diversity. Virology 2019, 539, 69–79. [Google Scholar] [CrossRef]
- Bijvand, Y.; Aghasadeghi, M.R.; Sakhaee, F.; Pakzad, P.; Vaziri, F.; Saraji, A.A.; Jamnani, F.R.; Siadat, S.D.; Fateh, A. First detection of human hepegivirus-1 (HHpgV-1) in Iranian patients with hemophilia. Sci. Rep. 2018, 8, 5036. [Google Scholar] [CrossRef]
- Wang, H.; Wan, Z.; Sun, Q.; Zhu, N.; Li, T.; Ren, X.; An, X.; Deng, S.; Wu, Y.; Li, X.; et al. Second human pegivirus in hepatitis C virus–infected and Hepatitis C virus/HIV-1–co-infected persons who inject drugs, China. Emerg. Infect. Dis. 2018, 24, 908–911. [Google Scholar] [CrossRef]
- Rodgers, M.A.; Holzmayer, V.; Vallari, A.; Olivo, A.; Forberg, K.; Fuhrman, J.; Coller, K.E.; Awazi, B.; Sidje, J.B.K.; Frankel, M.B.; et al. Hepatitis C virus surveillance and identification of human pegivirus 2 in a large Cameroonian cohort. J. Viral Hepat. 2018, 26, 30–37. [Google Scholar] [CrossRef]
- Scheel, T.K.; Simmonds, P.; Kapoor, A. Surveying the global virome: Identification and characterization of HCV-related animal hepaciviruses. Antivir. Res. 2015, 115, 83–93. [Google Scholar] [CrossRef]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2013, 59, 318–327. [Google Scholar] [CrossRef]
- Samadi, M.; Salimi, V.; Haghshenas, M.R.; Miri, S.M.; Mohebbi, S.R.; Ghaemi, A. Clinical and molecular aspects of human pegiviruses in the interaction host and infectious agent. Virol. J. 2022, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wan, Z.; Wang, J.H.; Yang, X.; Zhang, C. Review of human pegivirus: Prevalence, transmission, pathogenesis, and clinical implication. Virulence 2022, 13, 324–341. [Google Scholar] [CrossRef] [PubMed]
- Coller, K.E.; Bruce, V.; Cassidy, M.; Gersch, J.; Frankel, M.B.; Vallari, A.; Cloherty, G.; Hackett, J.; Evans, J.L.; Page, K.; et al. Chronic human Pegivirus 2 without Hepatitis C virus co-infection. Emerg. Infect. Dis. 2020, 26, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Coller, K.E.; Berg, M.G.; Frankel, M.; Forberg, K.; Surani, R.; Chiu, C.Y.; Hackett, J.; Dawson, G.J. Antibodies to the novel human Pegivirus 2 are associated with active and resolved infections. J. Clin. Microbiol. 2016, 54, 2023–2030. [Google Scholar] [CrossRef]
- Chivero, E.T.; Bhattarai, N.; Rydze, R.T.; Winters, M.A.; Holodniy, M.; Stapleton, J.T. Human pegivirus RNA is found in multiple blood mononuclear cells in vivo and serum-derived viral RNA-containing particles are infectious in vitro. J. Gen. Virol. 2014, 95, 1307–1319. [Google Scholar] [CrossRef]
- Chivero, E.T.; Stapleton, J.T. Tropism of human pegivirus (formerly known as GB virus C/hepatitis G virus) and host immunomodulation: Insights into a highly successful viral infection. J. Gen. Virol. 2015, 96, 1521–1532. [Google Scholar] [CrossRef]
- Fama, A.; Xiang, J.; Link, B.K.; Allmer, C.; Klinzman, D.; Feldman, A.L.; Nowakowski, G.S.; Liebow, M.; Larson, M.C.; Maurer, M.J.; et al. Human Pegivirus infection and lymphoma risk and prognosis: A North American study. Br. J. Haematol. 2018, 182, 644–653. [Google Scholar] [CrossRef]
- Wan, Z.; Liu, J.; Hu, F.; Shui, J.; Li, L.; Wang, H.; Tang, X.; Hu, C.; Liang, Y.; Zhou, Y.; et al. Evidence that the second human pegivirus (HPgV-2) is primarily a lymphotropic virus and can replicate independent of HCV replication. Emerg. Microbes Infect. 2020, 9, 485–495. [Google Scholar] [CrossRef]
- Liang, Y.; Hu, F.; Fan, H.; Li, L.; Wan, Z.; Wang, H.; Shui, J.; Zhou, Y.; Tong, Y.; Cai, W.; et al. Difference of intrahost dynamics of the second human pegivirus and hepatitis C virus in HPgV-2/HCV-coinfected patients. Front. Cell. Infect. Microbiol. 2021, 11, 728415. [Google Scholar] [CrossRef]
- Smith, E.C.; Sexton, N.R.; Denison, M.R. Thinking outside the triangle: Replication fidelity of the largest RNA viruses. Annu. Rev. Virol. 2014, 1, 111–132. [Google Scholar] [CrossRef]
- Ganai, R.A.; Johansson, E. DNA replication-a matter of fidelity. Mol. Cell 2016, 62, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, A.; Ziuzia-Graczyk, I. Fidelity of DNA replication-a matter of proofreading. Curr. Genet. 2018, 64, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef]
- Sanjuan, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life. Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S. Within-host viral diversity: A window into viral evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. The Evolution and Emergence of RNA Viruses; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005, 1, e11. [Google Scholar] [CrossRef]
- Cheung, P.H.P.; Watson, S.J.; Choy, K.-T.; Sia, S.F.; Wong, D.D.Y.; Poon, L.; Kellam, P.; Guan, Y.; Peiris, J.S.M.; Yen, H.-L. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Arnold, J.J.; Vignuzzi, M.; Stone, J.K.; Andino, R.; Cameron, C.E. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J. Biol. Chem. 2005, 280, 25706–25716. [Google Scholar] [CrossRef]
- Harrison, D.N.; Gazina, E.V.; Purcell, D.F.; Anderson, D.A.; Petrou, S. Amiloride derivatives inhibit coxsackievirus B3 RNA replication. J. Virol. 2008, 82, 1465–1473. [Google Scholar] [CrossRef][Green Version]
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Beeharry, Y.; Borderia, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar] [CrossRef] [PubMed]
- Gnädig, N.F.; Beaucourt, S.; Campagnola, G.; Bordería, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef]
- Liu, X.; Yang, X.; Lee, C.A.; Moustafa, I.M.; Smidansky, E.D.; Lum, D.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Vaccine-derived mutation in motif D of poliovirus RNA-dependent RNA polymerase lowers nucleotide incorporation fidelity. J. Biol. Chem. 2013, 288, 32753–32765. [Google Scholar] [CrossRef] [PubMed]
- Sadeghipour, S.; McMinn, P.C. A study of the virulence in mice of high copying fidelity variants of human enterovirus 71. Virus Res. 2013, 176, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wang, H.; Xie, X.; Yang, D.; Zhou, G.; Yu, L. An increased replication fidelity mutant of foot-and-mouth disease virus retains fitness in vitro and virulence in vivo. Antivir. Res. 2013, 100, 1–7. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, H.; Xie, X.; Li, C.; Zhou, G.; Yang, D.; Yu, L. Ribavirin-resistant variants of foot-and-mouth disease virus: The effect of restricted quasispecies diversity on viral virulence. J. Virol. 2014, 88, 4008–4020. [Google Scholar] [CrossRef]
- Rai, D.K.; Segundo, F.D.-S.; Campagnola, G.; Keith, A.; Schafer, E.A.; Kloc, A.; de los Santos, T.; Peersen, O.; Rieder, E. Attenuation of foot-and-mouth disease virus by engineered viral polymerase fidelity. J. Virol. 2017, 91, e00081-17. [Google Scholar] [CrossRef]
- Liu, W.; Shi, X.; Gong, P. A unique intra-molecular fidelity-modulating mechanism identified in a viral RNA-dependent RNA polymerase. Nucleic Acids Res. 2018, 46, 10840–10854. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Chu, J.T.S.; Wang, Y.; Chin, A.W.H.; Chong, T.H.; Dai, Z.; Poon, L.L.M.; Cheung, P.P.-H.; Huang, X. A novel mechanism of enhanced transcription activity and fidelity for influenza A viral RNA-dependent RNA polymerase. Nucleic Acids Res. 2021, 49, 8796–8810. [Google Scholar] [CrossRef]
- Furio, V.; Moya, A.; Sanjuan, R. The cost of replication fidelity in an RNA virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10233–10237. [Google Scholar] [CrossRef] [PubMed]
- Regoes, R.R.; Hamblin, S.; Tanaka, M.M. Viral mutation rates: Modelling the roles of within-host viral dynamics and the trade-off between replication fidelity and speed. Proc. Biol. Sci. 2013, 280, 20122047. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed–fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed]


 ) for Iran, CN (
) for Iran, CN ( ) for China, US (
) for China, US ( ) for the United States and VN (
) for the United States and VN ( ) for Vietnam.
) for Iran, CN () for China, US () for the United States and VN () for Vietnam.
) for Vietnam.
) for Iran, CN () for China, US () for the United States and VN () for Vietnam.

{kind=link}
{kind=link}
{kind=link}
| Groups | No. Tested | Anti-HPgV-2 (+) | HPgV-2 RNA (+) | ||
|---|---|---|---|---|---|
| China | Healthy Blood Donors | 4017 | 6 (0.15%) | 0 | |
| HBV (+) | 1000 | 2 (0.20%) | 0 | ||
| HIV-1 (+) | 539 | 0 | 0 | ||
| HCV (+) | 2440 | 30 (1.23%) | 7 (0.29%) | ||
| HCV (+)/HIV-1 (+) | 202 | 18 (8.91%) | 7 (3.47%) | ||
| Total | 8198 | 56 (0.68%) | 14 (0.17%) | ||
| United States | Healthy Blood Donors | 892 | 5 (0.56%) | 0 | |
| HBV (+) | 944 | 8 (0.85%) | 0 | ||
| HIV (+) | 928 | 5 (0.54%) | 0 | ||
| HCV (Ab+/NAT+) | 1708 | 46 (2.69%) | 22 (1.29%) | ||
| HCV (Ab+/NAT-) | 299 | 3 (1.00%) | 1 (0.33%) | ||
| Total | 4771 | 67 (1.40%) | 23 (0.48%) | ||
| Vietnam | Healthy Blood Donors | 80 | - | 0 | |
| HBV (+) | 103 | - | 0 | ||
| HIV (+) | 78 | - | 0 | ||
| HCV (+) | 394 | - | 0 | ||
| HCV (+)/HIV-1 (+) | 79 | - | 5 (6.3%) | ||
| Total | 734 | - | 5 (0.68%) | ||
| United Kingdom | Health Blood Donors | 50 | - | 0 | |
| Hemophilia | 195 | - | 1 (0.5%) | ||
| HIV (+) | 36 | - | 0 | ||
| HIV (+)/HCV (+)/PWID | 30 | - | 0 | ||
| HCV (+)/PWID | 120 | - | 2 (1.7%) | ||
| Total | 431 | - | 3 (0.7%) | ||
| Iran | Hemophilia patients (n = 436) | ||||
| HCV RNA (+) | HPgV-1 RNA (+) | TTV RNA (+) | HPgV-2 RNA (+) | HPgV-2 RNA (+)/HCV RNA (+)/HPgV-1 RNA (+)/TTV RNA (+) | |
| 163 (37.4%) | 19 (4.4%) | 76 (17.6%) | 4 (0.9%) | 4 (0.9%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Wang, H.; Dzakah, E.E.; Rashid, F.; Wang, J.; Tang, S. The Second Human Pegivirus, a Non-Pathogenic RNA Virus with Low Prevalence and Minimal Genetic Diversity. Viruses 2022, 14, 1844. https://doi.org/10.3390/v14091844
Chen S, Wang H, Dzakah EE, Rashid F, Wang J, Tang S. The Second Human Pegivirus, a Non-Pathogenic RNA Virus with Low Prevalence and Minimal Genetic Diversity. Viruses. 2022; 14(9):1844. https://doi.org/10.3390/v14091844
Chicago/Turabian StyleChen, Shuyi, Haiying Wang, Emmanuel Enoch Dzakah, Farooq Rashid, Jufang Wang, and Shixing Tang. 2022. "The Second Human Pegivirus, a Non-Pathogenic RNA Virus with Low Prevalence and Minimal Genetic Diversity" Viruses 14, no. 9: 1844. https://doi.org/10.3390/v14091844
APA StyleChen, S., Wang, H., Dzakah, E. E., Rashid, F., Wang, J., & Tang, S. (2022). The Second Human Pegivirus, a Non-Pathogenic RNA Virus with Low Prevalence and Minimal Genetic Diversity. Viruses, 14(9), 1844. https://doi.org/10.3390/v14091844