
Bunyaviral N Proteins Localize at RNA Processing Bodies and Stress Granules: The Enigma of Cytoplasmic Sources of Capped RNA for Cap Snatching

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of the Constructs
2.2. pEAQ-GFP (Modified), pEAQ-nsGFP and pEAQ-L-GFP Construction
2.3. Cell Culture, Transfection and Virus Infection
2.4. Immunostaining
2.5. Plant Material, Transient Expression and Virus Inoculation
2.6. Confocal Microscopy and Green Fluorescence Microscopy Observation
2.7. Quantification and Statistical Analysis
2.8. RNA Isolation and cDNA Synthesis
2.9. Quantitative Real-Time PCR (qRT-PCR)
3. Results
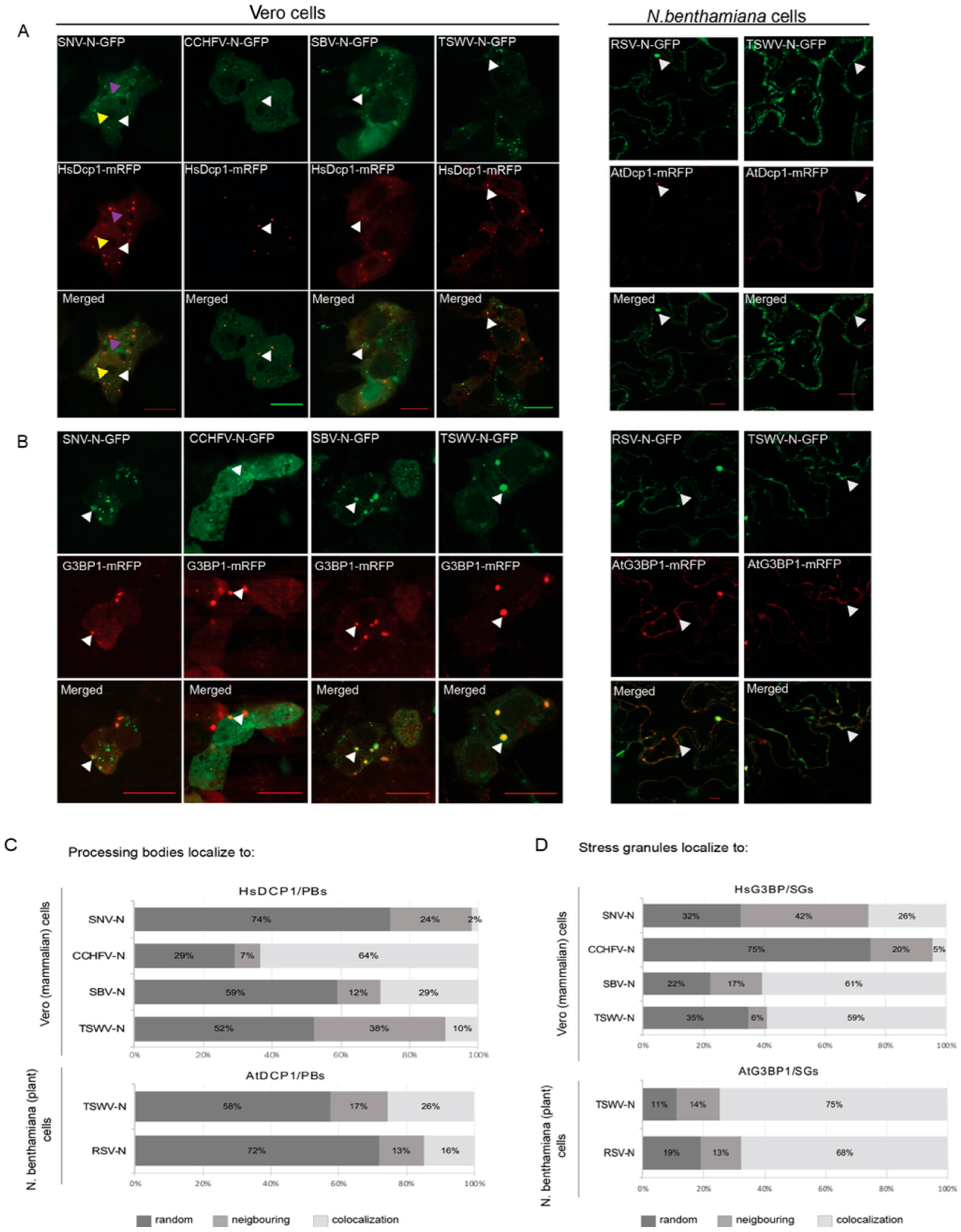
3.1. Viral N Proteins from Plant- and Animal-Infecting Members of the Bunyavirales Localize at PBs and SGs
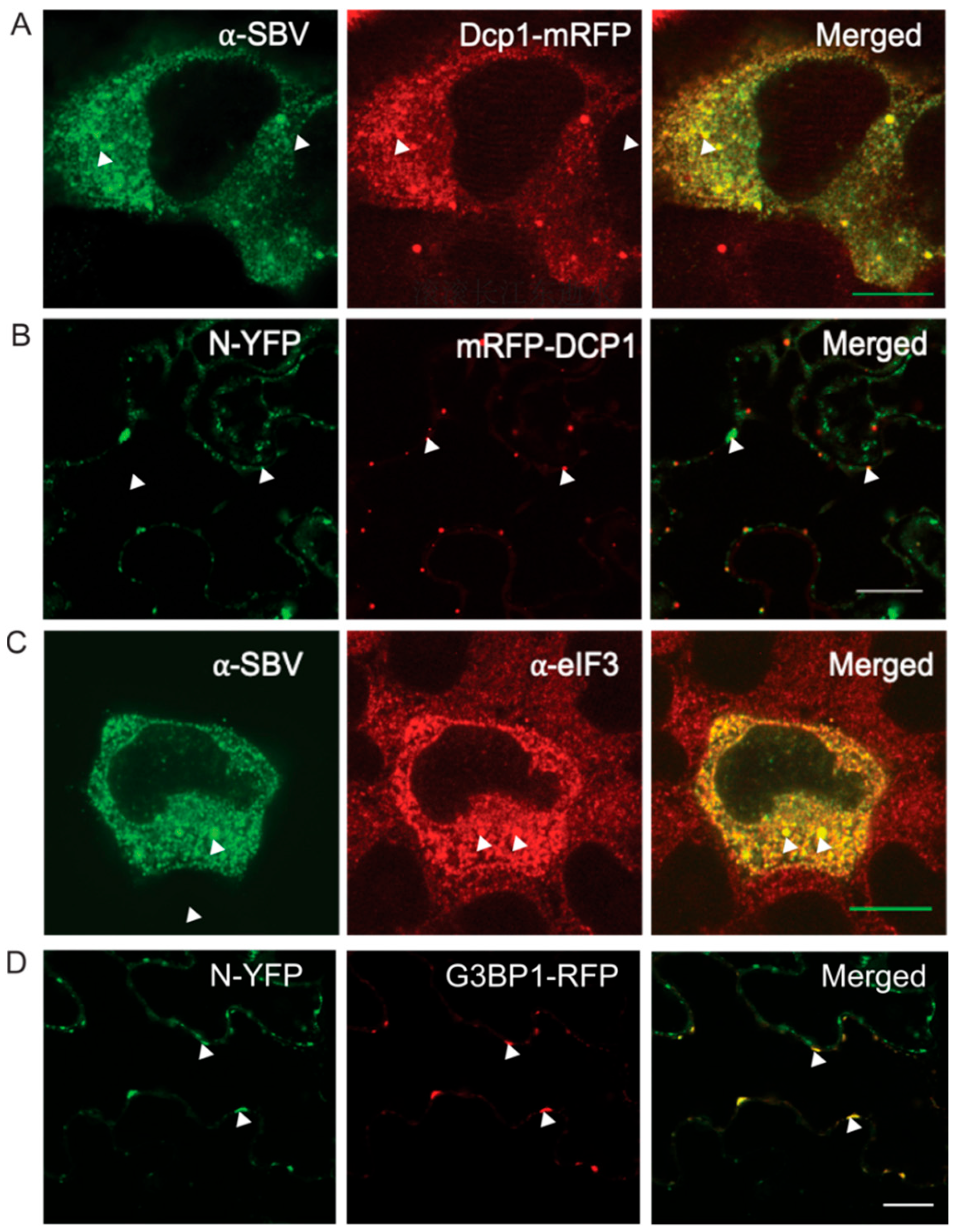
3.2. N Protein P Body/SG Association during Viral Infection
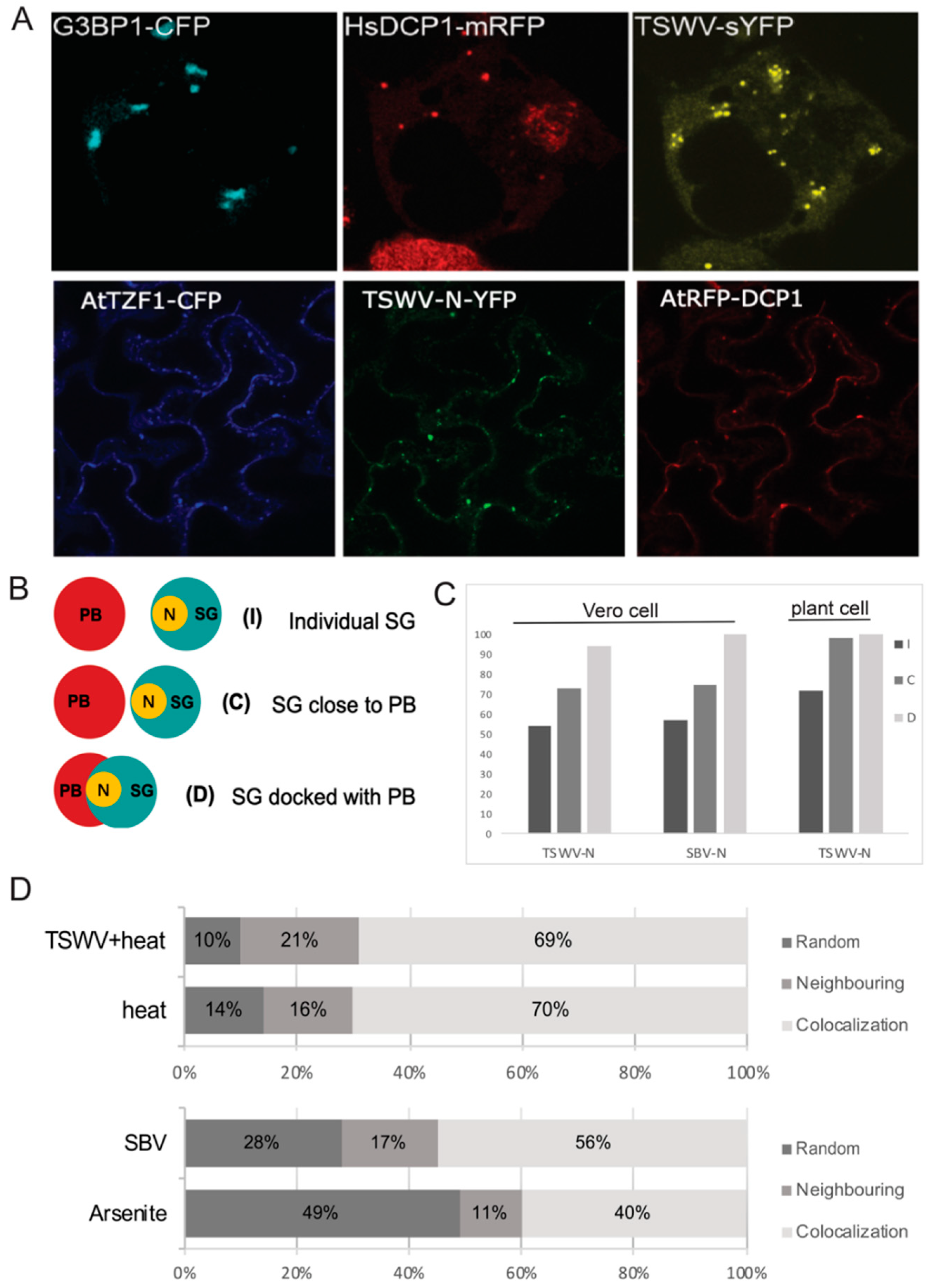
3.3. TSWV N Protein Preferably Localizes to SG Docked on P-Bodies, and the Formation of SG Docked on PB Is Promoted by Viral Infection
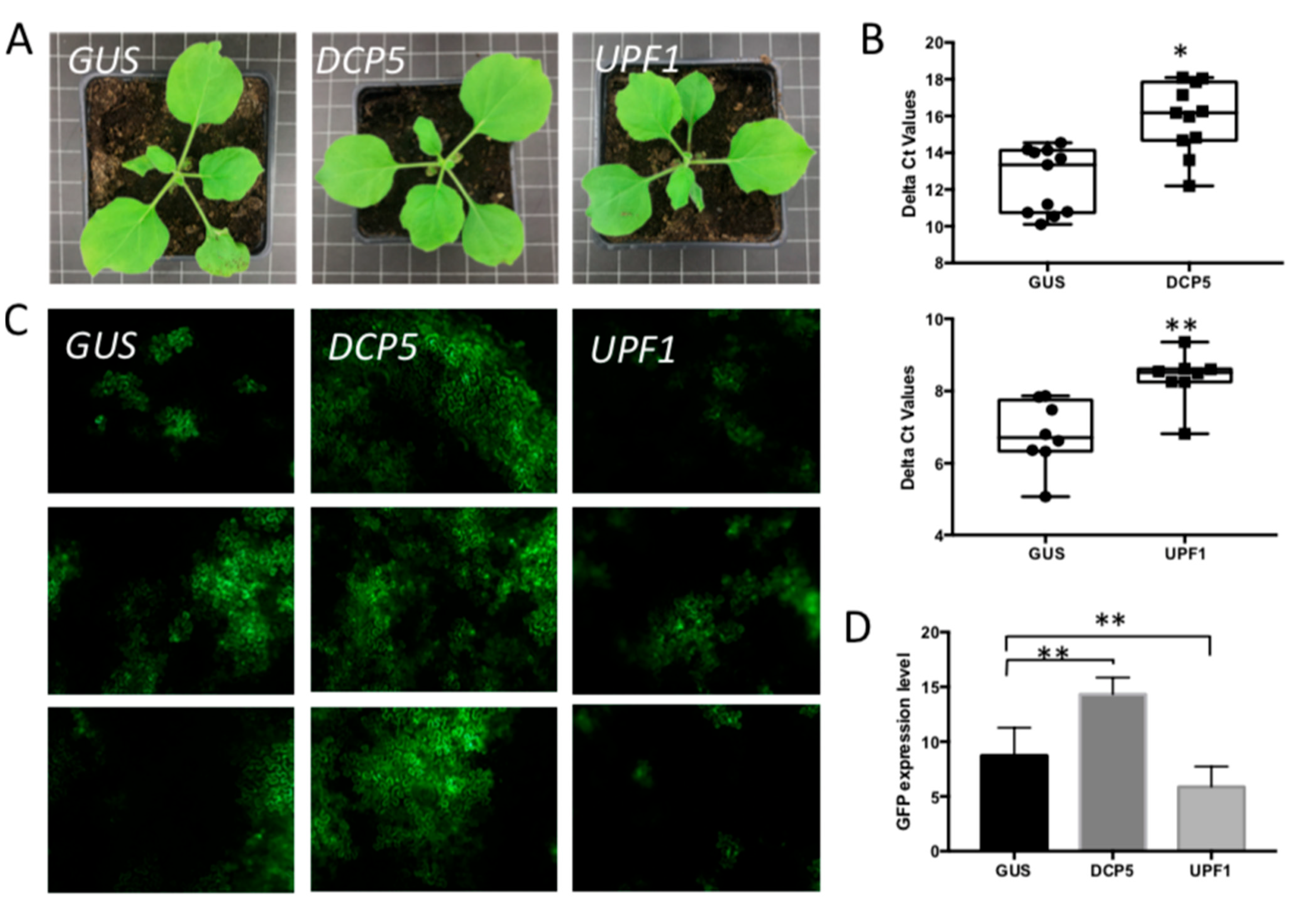
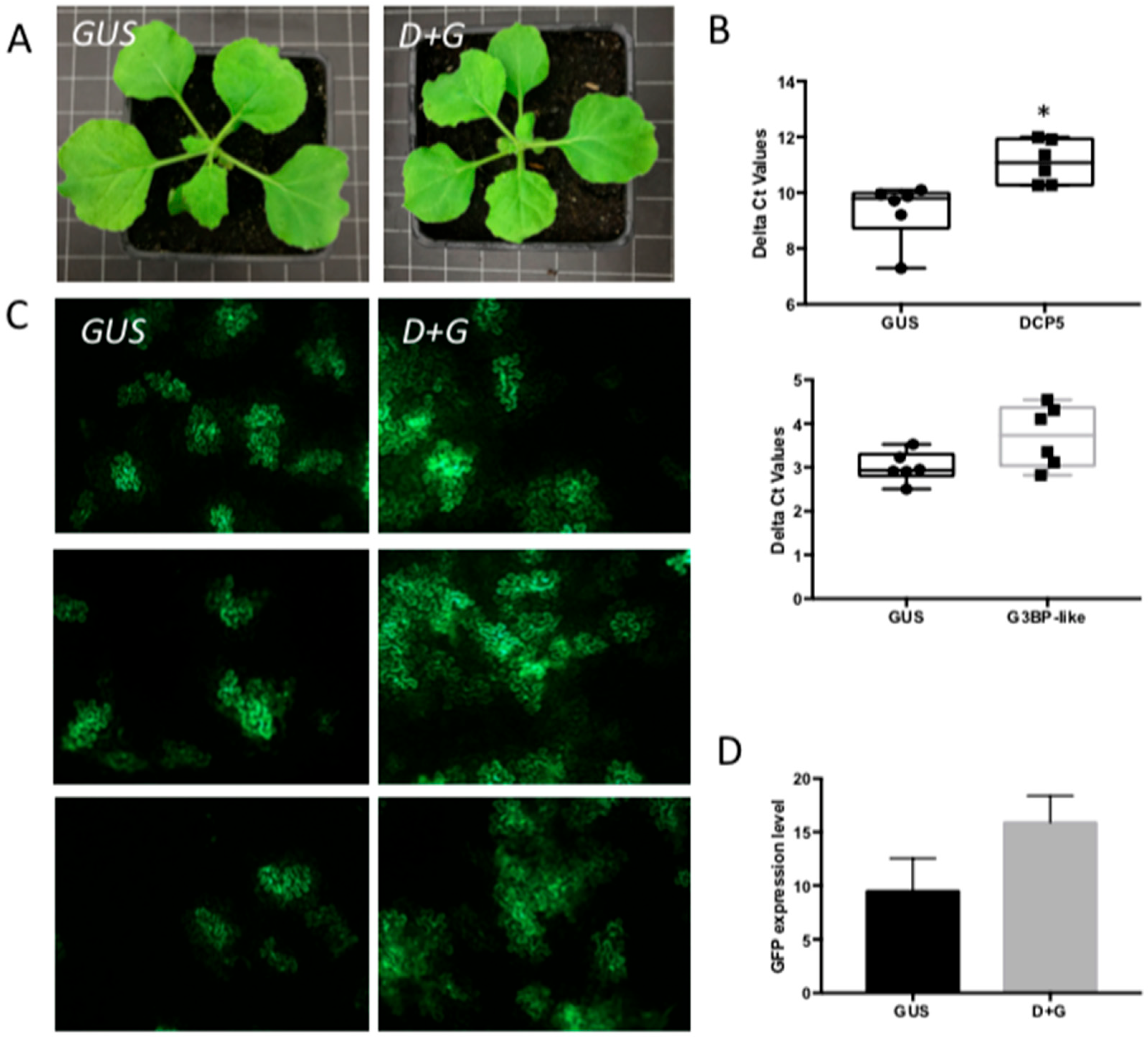
3.4. Silencing of Different PB- and SG-Resident Elements Differentially Influences TSWV Replication Rates
3.5. Viral Replication in Plants Simultaneously Silenced on PB-Resident DCP5 and SG-Resident G3BP1-like
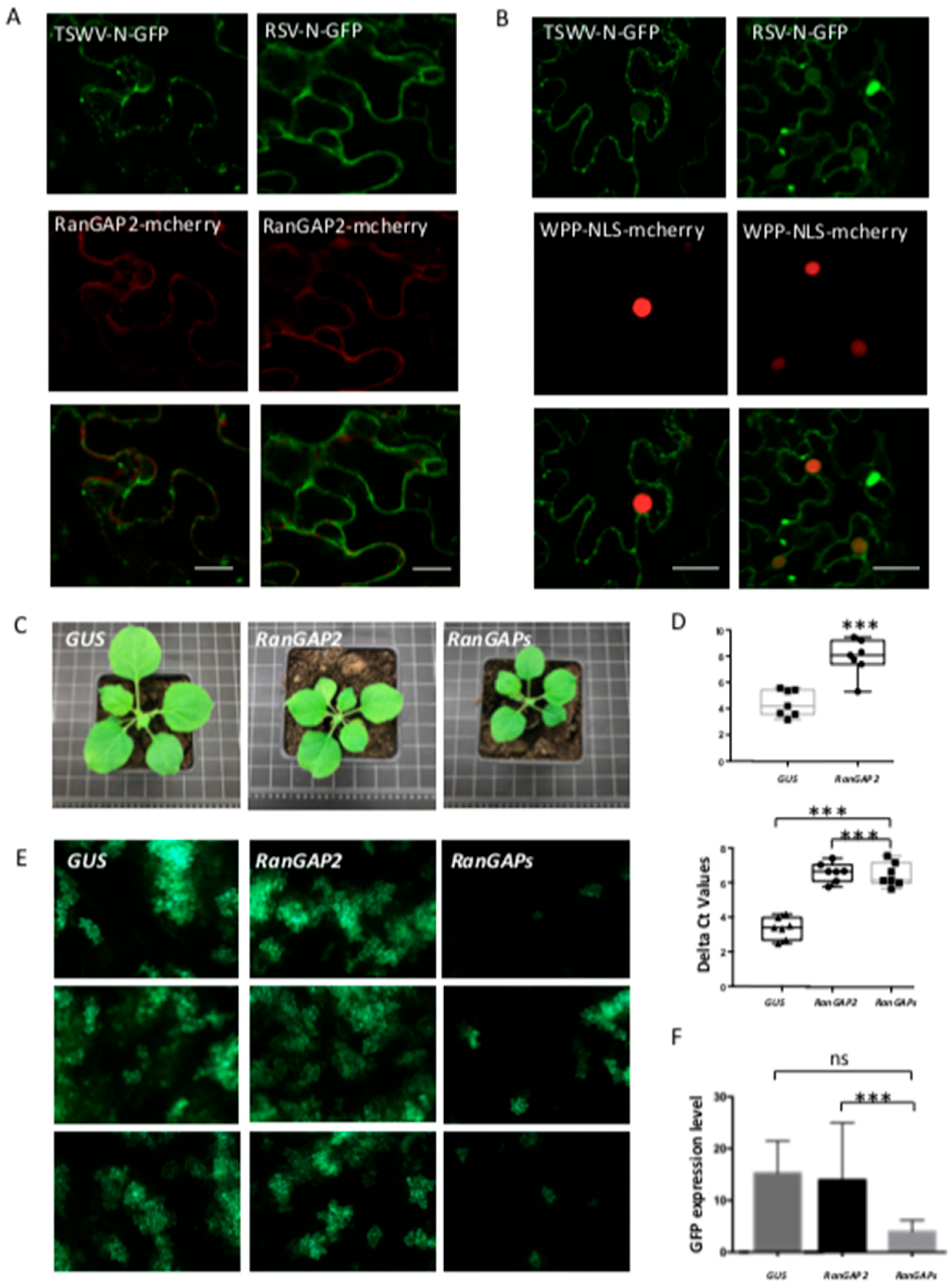
3.6. N Proteins Localize to the Perinuclear Region and Silencing of Nucleocytoplasmic Transport RanGAP Factors Negatively Affects TSWV Replication
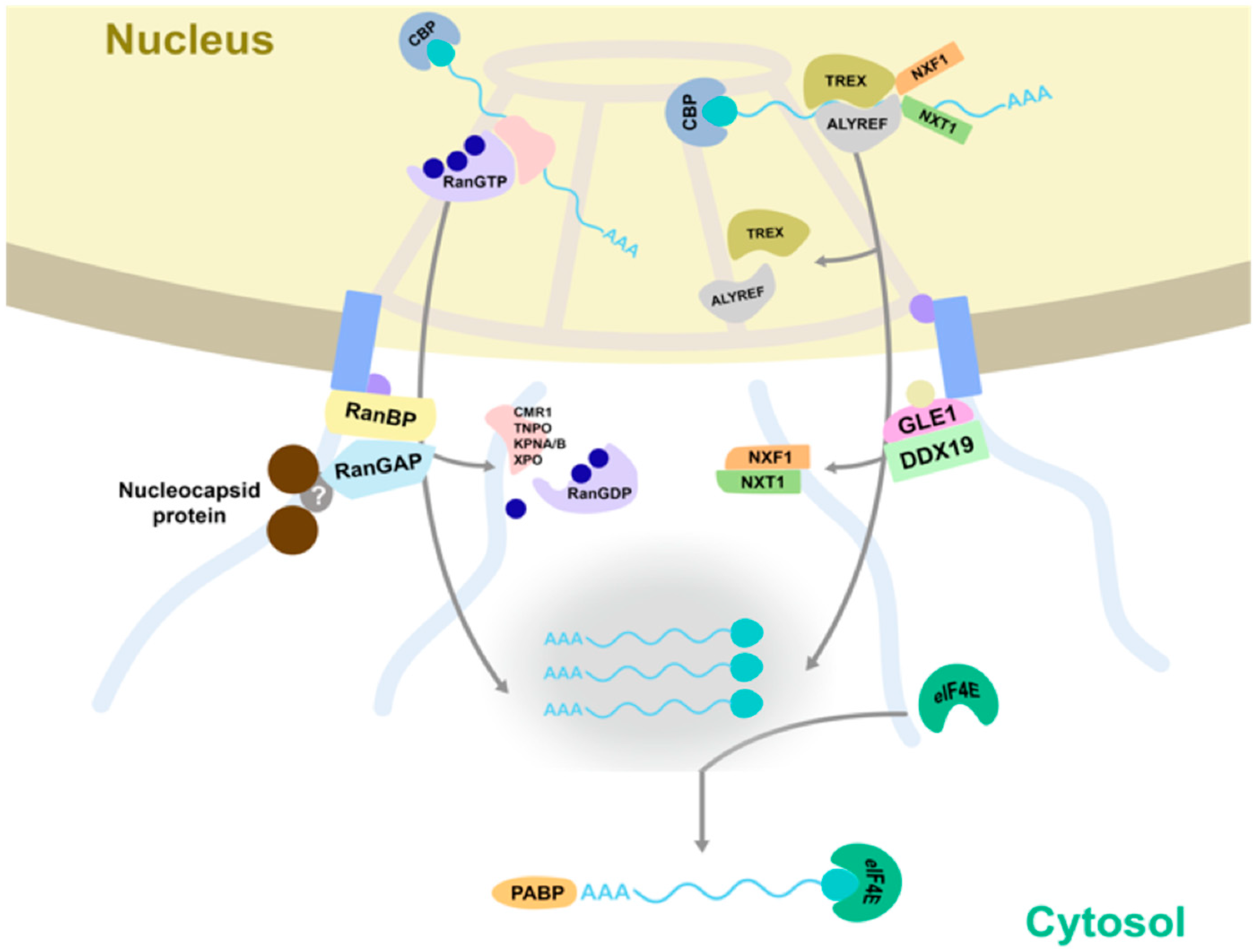
3.7. TSWV Cleaves Transcripts That End up at Different Locations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011, 10, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Gerlach, P.; Cusack, S. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr. Opin. Struct. Biol. 2016, 36, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crepin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Bartlam, M.; Lou, Z.; Chen, S.; Zhou, J.; He, X.; Lv, Z.; Ge, R.; Li, X.; Deng, T.; et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef]
- Guilligay, D.; Tarendeau, F.; Infante, P.R.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.H.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Stevaert, A.; Naesens, L. The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design. Med. Res. Rev. 2016, 36, 1127–1173. [Google Scholar] [CrossRef] [Green Version]
- Reguera, J.; Weber, F.; Cusack, S. Bunyaviridae RNA Polymerases (L-Protein) Have an N-Terminal, Influenza-Like Endonuclease Domain, Essential for Viral Cap-Dependent Transcription. PLOS Pathog. 2010, 6, e1001101. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, G.; He, G.; Peng, Y.; Liang, C. Characterization of an endonuclease in rice stripe tenuivirus Pc1 in vitro. Virus Res. 2019, 260, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, P.; Malet, H.; Cusack, S.; Reguera, J. Structural Insights into Bunyavirus Replication and Its Regulation by the vRNA Promoter. Cell 2015, 161, 1267–1279. [Google Scholar] [CrossRef] [Green Version]
- Gogrefe, N.; Reindl, S.; Günther, S.; Rosenthal, M. Structure of a functional cap-binding domain in Rift Valley fever virus L protein. PLOS Pathog. 2019, 15, e1007829. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.; Rosenthal, M.; Gogrefe, N.; Reindl, S.; Günther, S. Biochemical characterization of the Lassa virus L protein. J. Biol. Chem. 2019, 294, 8088–8100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, M.; Gogrefe, N.; Vogel, D.; Reguera, J.; Rauschenberger, B.; Cusack, S.; Günther, S.; Reindl, S. Structural insights into reptarenavirus cap-snatching machinery. PLOS Pathog. 2017, 13, e1006400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duijsings, D.; Kormelink, R.; Goldbach, R. Alfalfa mosaic virus RNAs serve as cap donors for tomato spotted wilt virus transcription during coinfection of Nicotiana benthamiana. J. Virol. 1999, 73, 5172–5175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Knippenberg, I.; Goldbach, R.; Kormelink, R. Purified Tomato spotted wilt virus Particles Support Both Genome Replication and Transcription in Vitro. Virology 2002, 303, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geerts-Dimitriadou, C.; Goldbach, R.; Kormelink, R. Preferential use of RNA leader sequences during influenza A transcription initiation in vivo. Virology 2011, 409, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Geerts-Dimitriadou, C.; Zwart, M.P.; Goldbach, R.; Kormelink, R. Base-pairing promotes leader selection to prime in vitro influenza genome transcription. Virology 2011, 409, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Zhang, T.-Q.; Zhou, T.; Zhou, Y.; Zhou, X.; Tao, X. Repetitive prime-and-realign mechanism converts short capped RNA leaders into longer ones that may be more suitable for elongation during rice stripe virus transcription initiation. J. Gen. Virol. 2012, 93 Pt 1, 194–202. [Google Scholar] [CrossRef]
- Liu, X.; Xiong, G.; Qiu, P.; Du, Z.; Kormelink, R.; Zheng, L.; Zhang, J.; Ding, X.; Yang, L.; Zhang, S.; et al. Inherent properties not conserved in other tenuiviruses increase priming and realignment cycles during transcription of Rice stripe virus. Virology 2016, 496, 287–298. [Google Scholar] [CrossRef]
- Cheng, E.; Mir, M.A. Signatures of Host mRNA 5′ Terminus for Efficient Hantavirus Cap Snatching. J. Virol. 2012, 86, 10173–10185. [Google Scholar] [CrossRef] [Green Version]
- Duijsings, D.; Kormelink, R.; Goldbach, R. In vivo analysis of the TSWV cap-snatching mechanism: Single base complementarity and primer length requirements. EMBO J. 2001, 20, 2545–2552. [Google Scholar] [CrossRef] [Green Version]
- van Knippenberg, I.; Lamine, M.; Goldbach, R.; Kormelink, R. Tomato spotted wilt virus transcriptase in vitro displays a preference for cap donors with multiple base complementarity to the viral template. Virology 2005, 335, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.Y.; Vreede, F.T.; Smith, M.; Engelhardt, O.G.; Fodor, E. Influenza virus inhibits RNA polymerase II elongation. Virology 2006, 351, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, O.G.; Smith, M.; Fodor, E. Association of the Influenza A Virus RNA-Dependent RNA Polymerase with Cellular RNA Polymerase II. J. Virol. 2005, 79, 5812–5818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olschewski, S.; Cusack, S.; Rosenthal, M. The Cap-Snatching Mechanism of Bunyaviruses. Trends Microbiol. 2020, 28, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-C.; Lloyd, R.E. Cytoplasmic RNA Granules and Viral Infection. Annu. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [Green Version]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Parker, R.; Sheth, U. P Bodies and the Control of mRNA Translation and Degradation. Mol. Cell 2007, 25, 635–646. [Google Scholar] [CrossRef]
- Beckham, C.J.; Parker, R. P Bodies, Stress Granules, and Viral Life Cycles. Cell Host Microbe 2008, 3, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Malinowska, M.; Niedźwiedzka-Rystwej, P.; Tokarz-Deptuła, B.; Deptuła, W. Stress granules (SG) and processing bodies (PB) in viral infections. Acta Biochim. Pol. 2016, 63, 183–188. [Google Scholar] [CrossRef]
- Mäkinen, K.; Lõhmus, A.; Pollari, M. Plant RNA Regulatory Network and RNA Granules in Virus Infection. Front. Plant Sci. 2017, 8, 2093. [Google Scholar] [CrossRef] [Green Version]
- Krapp, S.; Greiner, E.; Amin, B.; Sonnewald, U.; Krenz, B. The stress granule component G3BP is a novel interaction partner for the nuclear shuttle proteins of the nanovirus pea necrotic yellow dwarf virus and geminivirus abutilon mosaic virus. Virus Res. 2017, 227, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Mazur, M.J.; Tao, X.; Kormelink, R. Cellular RNA Hubs: Friends and Foes of Plant Viruses. Mol. Plant-Microbe Interact. 2020, 33, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.A.; Duran, W.A.; Hjelle, B.L.; Ye, C.; Panganiban, A.T. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc. Natl. Acad. Sci. USA 2008, 105, 19294–19299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, A.; Mir, M.A. Interaction of Hantavirus Nucleocapsid Protein with Ribosomal Protein S19. J. Virol. 2010, 84, 12450–12453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panganiban, A.T.; Mir, M.A. Bunyavirus N: eIF4F surrogate and cap-guardian. Cell Cycle 2009, 8, 1332–1337. [Google Scholar] [CrossRef] [Green Version]
- Linero, F.; Welnowska, E.; Carrasco, L.; Scolaro, L. Participation of eIF4F complex in Junin virus infection: Blockage of eIF4E does not impair virus replication. Cell Microbiol. 2013, 15, 1766–1782. [Google Scholar] [CrossRef]
- Geerts-Dimitriadou, C.; Lu, Y.-Y.; Geertsema, C.; Goldbach, R.; Kormelink, R. Analysis of the Tomato spotted wilt virus Ambisense S RNA-Encoded Hairpin Structure in Translation. PLoS ONE 2012, 7, e31013. [Google Scholar] [CrossRef]
- Jeeva, S.; Cheng, E.; Ganaie, S.S.; Mir, M.A. Crimean-Congo Hemorrhagic Fever Virus Nucleocapsid Protein Augments mRNA Translation. J. Virol. 2017, 91, e00636-17. [Google Scholar] [CrossRef] [Green Version]
- Jeeva, S.; Pador, S.; Voss, B.; Ganaie, S.S.; Mir, M.A. Crimean-Congo hemorrhagic fever virus nucleocapsid protein has dual RNA binding modes. PLoS ONE 2017, 12, e0184935. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhou, Y.; Moffett, P. Alterations in cellular RNA decapping dynamics affect tomato spotted wilt virus cap snatching and infection in Arabidopsis. New Phytol. 2019, 224, 789–803. [Google Scholar] [CrossRef]
- Dubin, M.J.; Bowler, C.; Benvenuto, G. A modified Gateway cloning strategy for overexpressing tagged proteins in plants. Plant Methods 2008, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainsbury, F.; Thuenemann, E.C.; Lomonossoff, G.P. pEAQ: Versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol. J. 2009, 7, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schiff, M.; Dinesh-Kumar, S.P. Virus-induced gene silencing in tomato. Plant J. 2002, 31, 777–786. [Google Scholar] [CrossRef]
- Feng, M.; Cheng, R.; Chen, M.; Guo, R.; Li, L.; Feng, Z.; Wu, J.; Xie, L.; Hong, J.; Zhang, Z.; et al. Rescue of tomato spotted wilt virus entirely from complementary DNA clones. Proc. Natl. Acad. Sci. USA 2020, 117, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Verschoor, A.; Kormelink, R.; Rottier, P.; Goldbach, R. Tomato Spotted Wilt Virus Glycoproteins Exhibit Trafficking and Localization Signals That Are Functional in Mammalian Cells. J. Virol. 2001, 75, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snippe, M.; Borst, J.W.; Goldbach, R.; Kormelink, R. Tomato spotted wilt virus Gc and N proteins interact in vivo. Virology 2007, 357, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snippe, M.; Borst, J.W.; Goldbach, R.; Kormelink, R. The use of fluorescence microscopy to visualise homotypic interactions of tomato spotted wilt virus nucleocapsid protein in living cells. J. Virol. Methods 2005, 125, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chua, N.-H. Arabidopsis Decapping 5 Is Required for mRNA Decapping, P-Body Formation, and Translational Repression during Postembryonic Development. Plant Cell 2009, 21, 3270–3279. [Google Scholar] [CrossRef] [Green Version]
- Chicois, C.; Scheer, H.; Garcia, S.; Zuber, H.; Mutterer, J.; Chicher, J.; Hammann, P.; Gagliardi, D.; Garcia, D. The UPF1 interactome reveals interaction networks between RNA degradation and translation repression factors in Arabidopsis. Plant J. 2018, 96, 119–132. [Google Scholar] [CrossRef]
- Kim, Y.K.; Maquat, L.E. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 2019, 25, 407–422. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell 2017, 68, 808–820.e5. [Google Scholar] [CrossRef]
- Hopkins, K.C.; McLane, L.M.; Maqbool, T.; Panda, D.; Gordesky-Gold, B.; Cherry, S. A genome-wide RNAi screen reveals that mRNA decapping restricts bunyaviral replication by limiting the pools of Dcp2-accessible targets for cap-snatching. Genes Dev. 2013, 27, 1511–1525. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Nover, L.; Fauth, M. Plant stress granules and mRNA processing bodies are distinct from heat stress granules. Plant J. 2008, 56, 517–530. [Google Scholar] [CrossRef]
- Mir, M.A.; Sheema, S.; Haseeb, A.; Haque, A. Hantavirus Nucleocapsid Protein Has Distinct m7G Cap- and RNA-binding Sites. J. Biol. Chem. 2010, 285, 11357–11368. [Google Scholar] [CrossRef] [Green Version]
- Bellocq, C.; Kolakofsky, D. Translational requirement for La Crosse virus S-mRNA synthesis: A possible mechanism. J. Virol. 1987, 61, 3960–3967. [Google Scholar] [CrossRef] [Green Version]
- Vialat, P.; Bouloy, M. Germiston virus transcriptase requires active 40S ribosomal subunits and utilizes capped cellular RNAs. J. Virol. 1992, 66, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Sulkowska, A.; Auber, A.; Sikorski, P.J.; Auth, M.; Sitkiewicz, E.; Jean, V.; Kufel, J. RNA Helicases from the DEA(D/H)-Box Family Contribute to Plant NMD Efficiency. Plant Cell Physiol. 2020, 61, 144–157. [Google Scholar] [CrossRef]
- Gamage, S.W.; Dietzgen, R.G. Intracellular Localization, Interactions and Functions of Capsicum Chlorosis Virus Proteins. Front. Microbiol. 2017, 8, 612. [Google Scholar] [CrossRef] [Green Version]
- Ravkov, E.V.; Compans, R.W. Hantavirus Nucleocapsid Protein Is Expressed as a Membrane-Associated Protein in the Perinuclear Region. J. Virol. 2001, 75, 1808–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichelt, M.; Stertz, S.; Krijnse-Locker, J.; Haller, O.; Kochs, G. Missorting of LaCrosse Virus Nucleocapsid Protein by the Interferon-Induced MxA GTPase Involves Smooth ER Membranes. Traffic 2004, 5, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.; Bladh, L.; Mousavi-Jazi, M.; Magnusson, K.-E.; Lundkvist, A.; Haller, O.; Mirazimi, A. Human MxA Protein Inhibits the Replication of Crimean-Congo Hemorrhagic Fever Virus. J. Virol. 2004, 78, 4323–4329. [Google Scholar] [CrossRef] [Green Version]
- Andersson, I.; Simon, M.; Lundkvist, A.; Nilsson, M.; Holmström, A.; Elgh, F.; Mirazimi, A. Role of actin filaments in targeting of Crimean Congo hemorrhagic fever virus nucleocapsid protein to perinuclear regions of mammalian cells. J. Med. Virol. 2004, 72, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Zilman, A.; Di Talia, S.; Chait, B.T.; Rout, M.P.; Magnasco, M.O. Efficiency, Selectivity, and Robustness of Nucleocytoplasmic Transport. PLOS Comput. Biol. 2007, 3, e125. [Google Scholar] [CrossRef] [Green Version]
- Fiserova, J.; Spink, M.; Richards, S.; Saunter, C.; Goldberg, M. Entry into the nuclear pore complex is controlled by a cytoplasmic exclusion zone containing dynamic GLFG-repeat nucleoporin domains. J. Cell Sci. 2013, 127 Pt 1, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Lui, K.; Huang, Y. RanGTPase: A Key Regulator of Nucleocytoplasmic Trafficking. Mol. Cell. Pharmacol. 2009, 1, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Remenyi, R.; Gao, Y.; Hughes, R.E.; Curd, A.; Zothner, C.; Peckham, M.; Merits, A.; Harris, M. Persistent Replication of a Chikungunya Virus Replicon in Human Cells Is Associated with Presence of Stable Cytoplasmic Granules Containing Nonstructural Protein 3. J. Virol. 2018, 92, e00477-18. [Google Scholar] [CrossRef] [Green Version]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus Infection Induces Discrete Cytosolic Structures for RNA Replication. J. Virol. 2012, 86, 11301–11310. [Google Scholar] [CrossRef] [Green Version]
- Haimovich, G.; Choder, M.; Singer, R.H.; Trcek, T. The fate of the messenger is pre-determined: A new model for regulation of gene expression. Biochim. Et Biophys. Acta 2013, 1829, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Sarkar, D.; Das, B. The interplay between transcription and mRNA degradation in Saccharomyces cerevisiae. Microb. Cell 2017, 4, 212–228. [Google Scholar] [CrossRef] [Green Version]
- Brune, C.; Munchel, S.E.; Fischer, N.; Podtelejnikov, A.V.; Weis, K. Yeast poly(A)-binding protein Pab1 shuttles between the nucleus and the cytoplasm and functions in mRNA export. RNA 2005, 11, 517–531. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Mazur, M.; Gulickx, N.; Hong, H.; Overmars, H.; Tao, X.; Kormelink, R. Bunyaviral N Proteins Localize at RNA Processing Bodies and Stress Granules: The Enigma of Cytoplasmic Sources of Capped RNA for Cap Snatching. Viruses 2022, 14, 1679. https://doi.org/10.3390/v14081679
Xu M, Mazur M, Gulickx N, Hong H, Overmars H, Tao X, Kormelink R. Bunyaviral N Proteins Localize at RNA Processing Bodies and Stress Granules: The Enigma of Cytoplasmic Sources of Capped RNA for Cap Snatching. Viruses. 2022; 14(8):1679. https://doi.org/10.3390/v14081679
Chicago/Turabian StyleXu, Min, Magdalena Mazur, Nigel Gulickx, Hao Hong, Hein Overmars, Xiaorong Tao, and Richard Kormelink. 2022. "Bunyaviral N Proteins Localize at RNA Processing Bodies and Stress Granules: The Enigma of Cytoplasmic Sources of Capped RNA for Cap Snatching" Viruses 14, no. 8: 1679. https://doi.org/10.3390/v14081679
APA StyleXu, M., Mazur, M., Gulickx, N., Hong, H., Overmars, H., Tao, X., & Kormelink, R. (2022). Bunyaviral N Proteins Localize at RNA Processing Bodies and Stress Granules: The Enigma of Cytoplasmic Sources of Capped RNA for Cap Snatching. Viruses, 14(8), 1679. https://doi.org/10.3390/v14081679