
The Impact of Epitranscriptomics on Antiviral Innate Immunity



Abstract
:1. Introduction
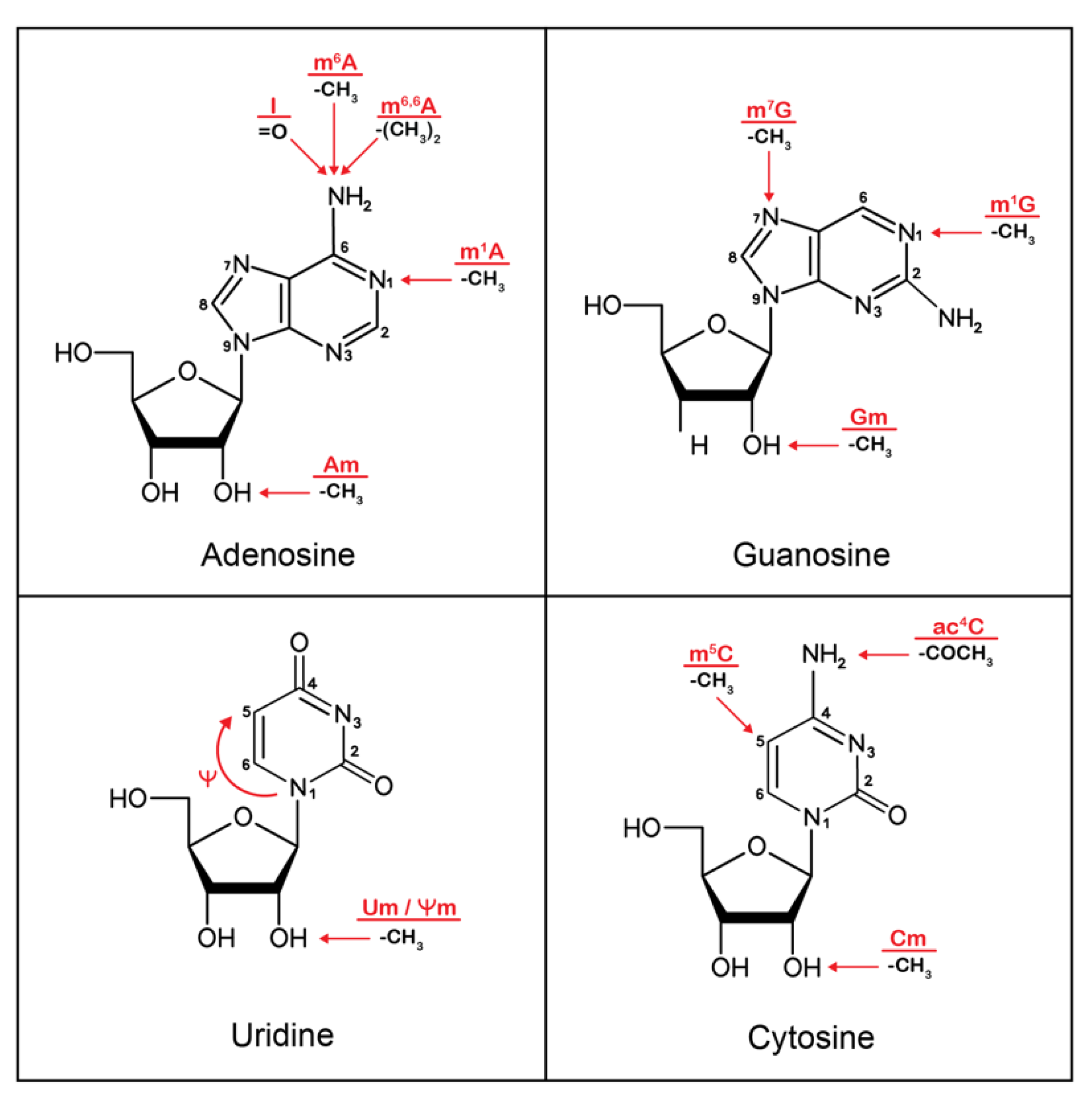
2. Epitranscriptomics
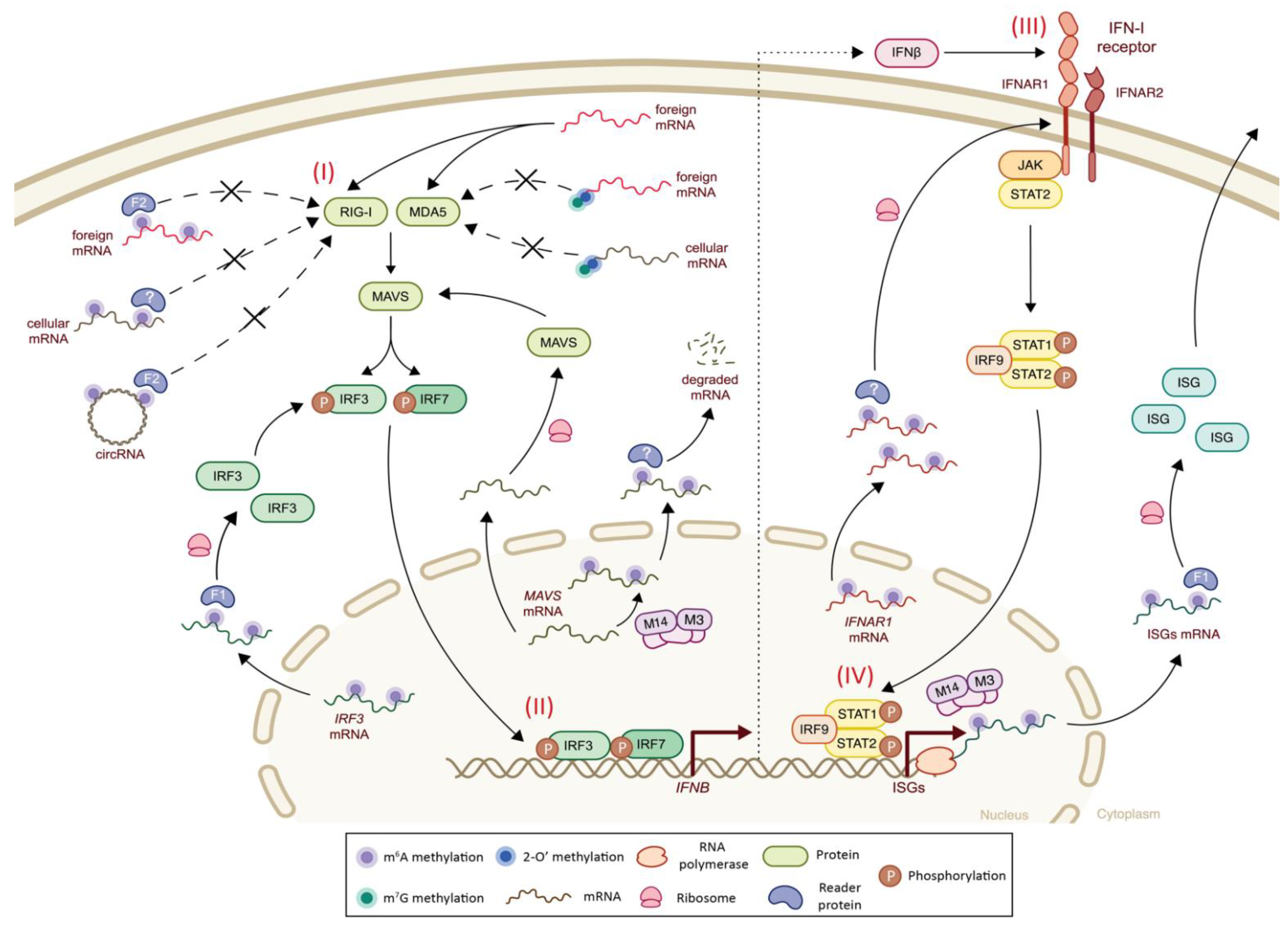
3. Epitranscriptomics and Innate Immunity
4. Viral Epitranscriptomics and Innate Immunity
5. RNA Viruses
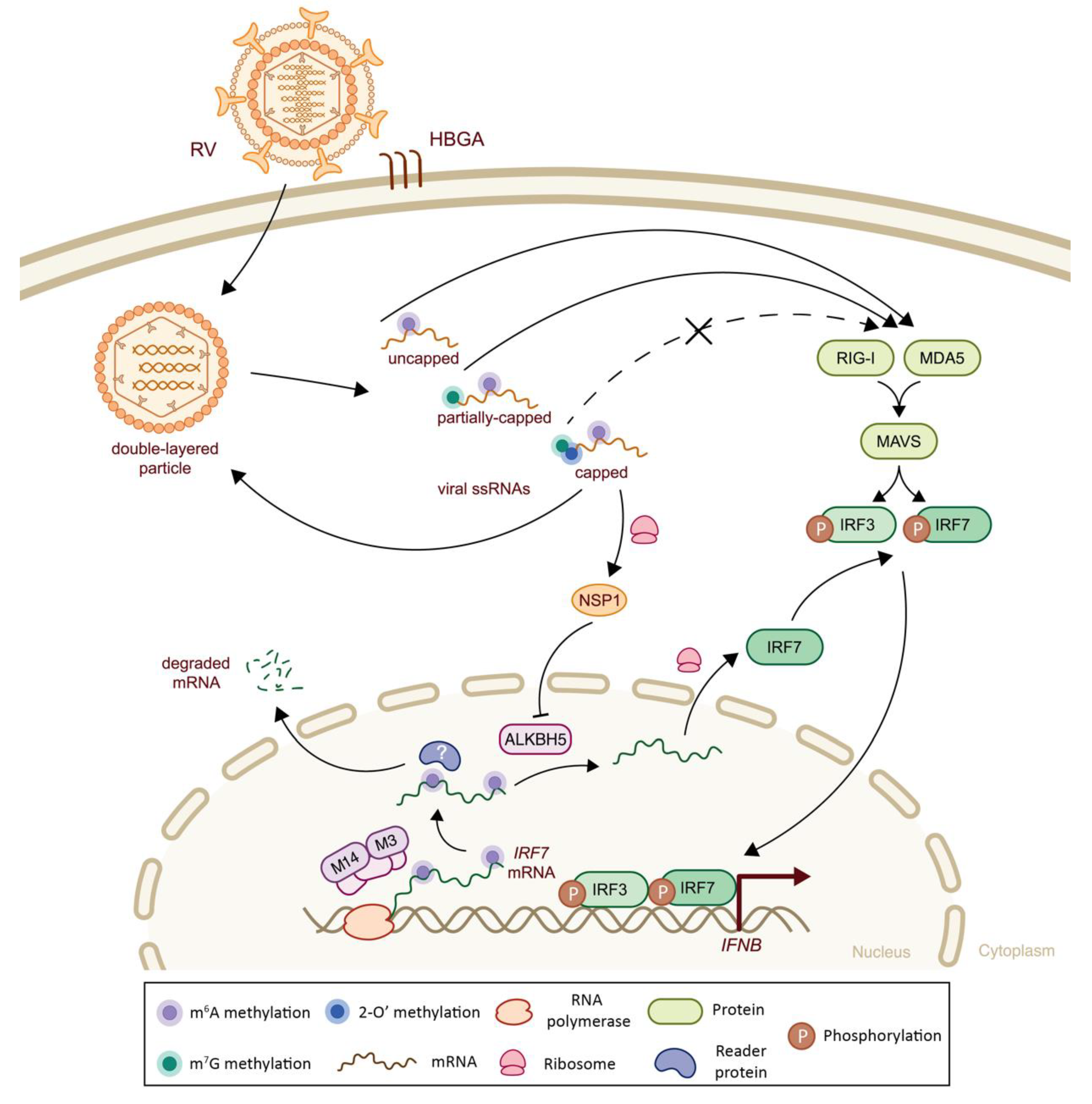
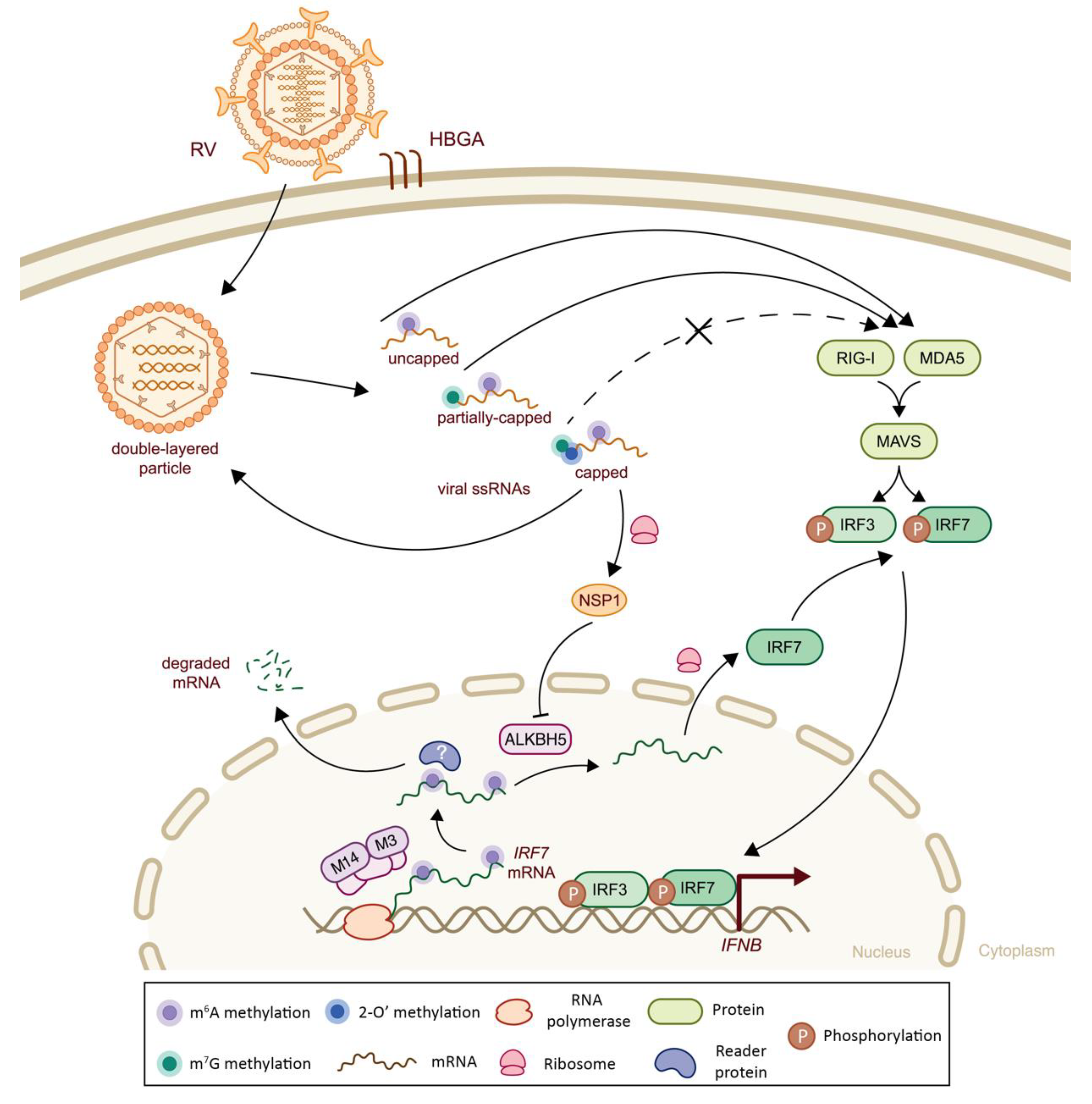
5.1. Class III Viruses: Double-Stranded RNA
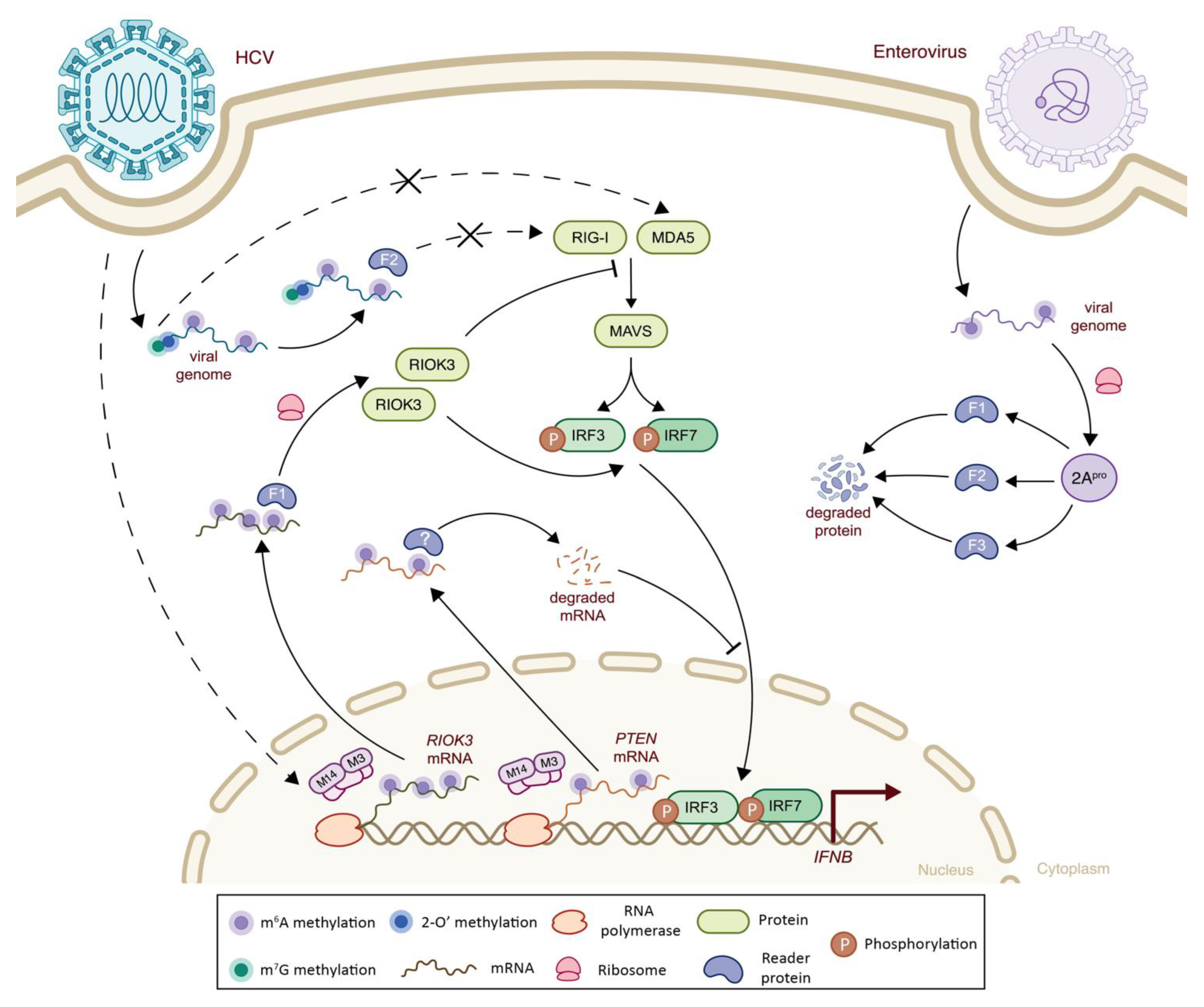
5.2. Class IV Viruses: Positive Sense, Single-Stranded RNA
5.3. Class V Viruses: Negative Sense, Single-Stranded RNA
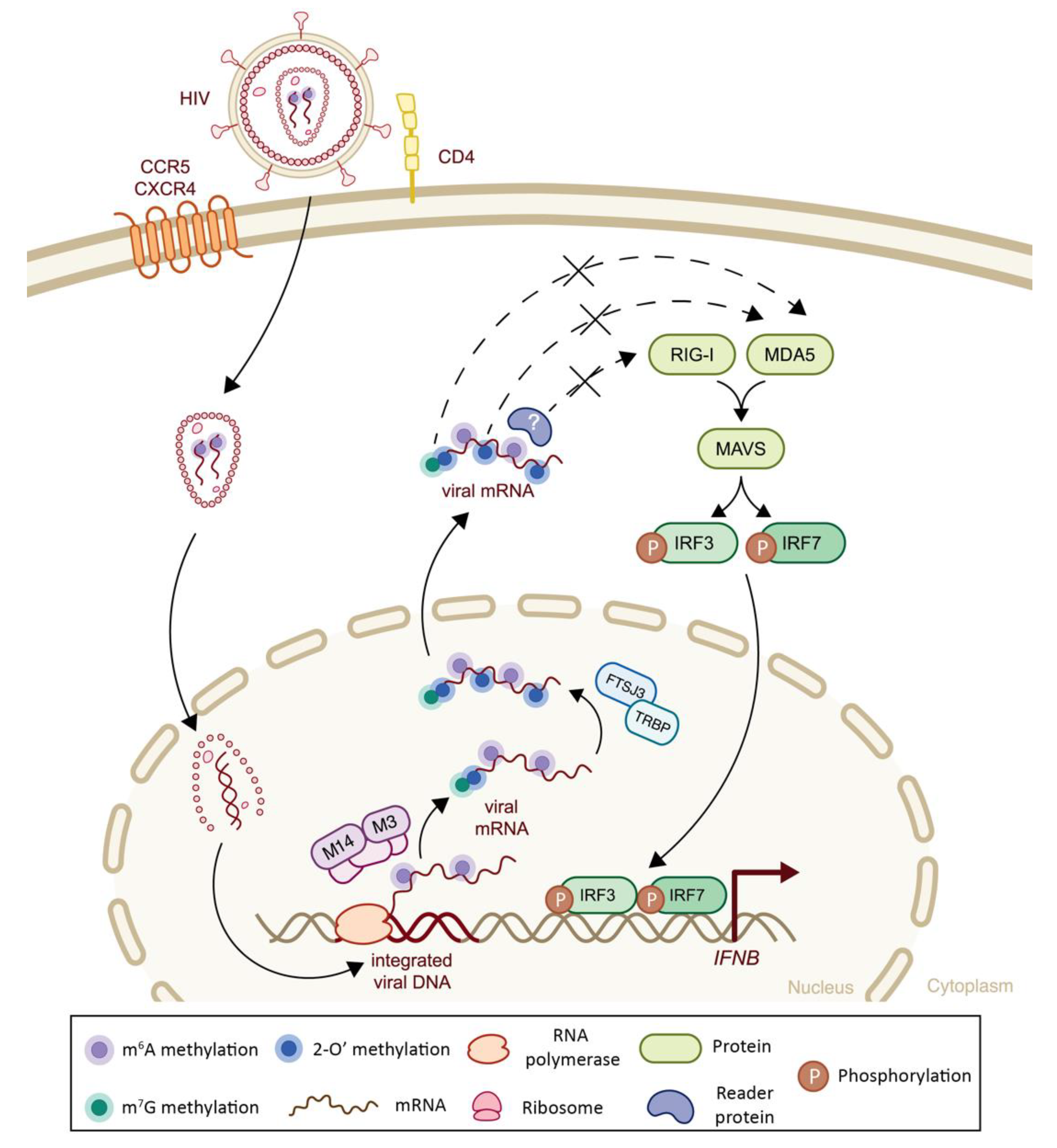
5.4. Class VI Viruses: Positive Sense, Single-Stranded RNA with Reverse Transcriptase
6. DNA Viruses
6.1. Class VII Viruses: Double-Stranded DNA with Rverse Transcriptase
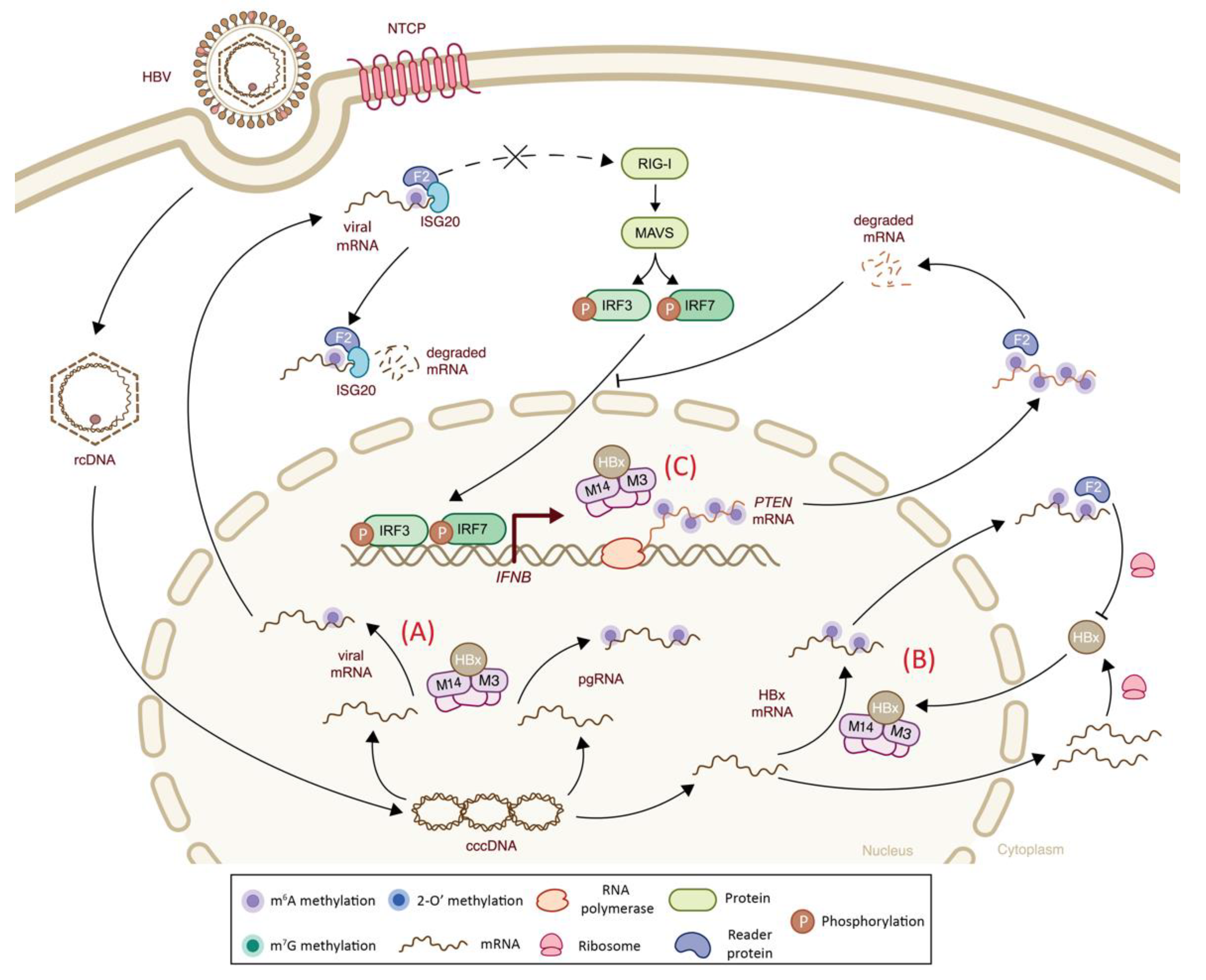
6.2. Class I Viruses: Double-Stranded DNA
6.3. Class II Viruses: Single-Stranded DNA
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davis, F.F.; Allen, F.W. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 1957, 227, 907–915. [Google Scholar] [CrossRef]
- Ozban, N.; Tandler, J.; Sirlin, J.L. Methylation of Nucleolar Rna during Development of the Amphibian Ooecyte. J. Embryol. Exp. Morphol. 1964, 12, 373–380. [Google Scholar] [PubMed]
- Edmonds, M.; Vaughan, M.H., Jr.; Nakazato, H. Polyadenylic acid sequences in the heterogeneous nuclear RNA and rapidly-labeled polyribosomal RNA of HeLa cells: Possible evidence for a precursor relationship. Proc. Natl. Acad. Sci. USA 1971, 68, 1336–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.A.; Paoletti, E.; Moss, B. Purification of mRNA guanylyltransferase and mRNA (guanine-7-) methyltransferase from vaccinia virions. J. Biol. Chem. 1975, 250, 9322–9329. [Google Scholar] [CrossRef]
- Dunin-Horkawicz, S.; Czerwoniec, A.; Gajda, M.J.; Feder, M.; Grosjean, H.; Bujnicki, J.M. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2006, 34, D145–D149. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Flores, M.A.; Meng, J.; Zhang, L.; Zhao, X.; Rao, M.K.; Chen, Y.; Huang, Y. MeT-DB: A database of transcriptome methylation in mammalian cells. Nucleic Acids Res. 2015, 43, D197–D203. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Davalos, V.; Blanco, S.; Esteller, M. SnapShot: Messenger RNA Modifications. Cell 2018, 174, 498–498.e1. [Google Scholar] [CrossRef]
- Nombela, P.; Miguel-Lopez, B.; Blanco, S. The role of m6A, m5C and Ψ RNA modifications in cancer: Novel therapeutic opportunities. Mol. Cancer 2021, 20, 18. [Google Scholar] [CrossRef]
- Song, B.; Chen, K.; Tang, Y.; Wei, Z.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J. ConsRM: Collection and large-scale prediction of the evolutionarily conserved RNA methylation sites, with implications for the functional epitranscriptome. Brief. Bioinform. 2021, 22, bbab088. [Google Scholar] [CrossRef] [PubMed]
- Selaru, A.; Costache, M.; Dinescu, S. Epitranscriptomic signatures in stem cell differentiation to the neuronal lineage. RNA Biol. 2021, 18, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Li, J.; Zhao, B.; Huang, G.; Li, X.; Xie, Z.; Zhou, Z. The Emerging Role of m6A Modification in Regulating the Immune System and Autoimmune Diseases. Front. Cell Dev. Biol. 2021, 9, 755691. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Courtney, D.G.; Tsai, K.; Cullen, B.R. Viral Epitranscriptomics. J. Virol. 2017, 91, e02263-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Roundtree, I.A.; Luo, G.Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef]
- Park, O.H.; Ha, H.; Lee, Y.; Boo, S.H.; Kwon, D.H.; Song, H.K.; Kim, Y.K. Endoribonucleolytic Cleavage of m6A-Containing RNAs by RNase P/MRP Complex. Mol. Cell 2019, 74, 494–507.e8. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. BioMed Res. Int. 2013, 2013, 683095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Li, X.; Qi, R.; Billiar, T. RNA Editing, ADAR1, and the Innate Immune Response. Genes 2017, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Lerner, T.; Papavasiliou, F.N.; Pecori, R. RNA Editors, Cofactors, and mRNA Targets: An Overview of the C-to-U RNA Editing Machinery and Its Implication in Human Disease. Genes 2018, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Nakahama, T.; Kawahara, Y. Adenosine-to-inosine RNA editing in the immune system: Friend or foe? Cell Mol. Life Sci. 2020, 77, 2931–2948. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- He, P.C.; He, C. m6 A RNA methylation: From mechanisms to therapeutic potential. EMBO J. 2021, 40, e105977. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N6-methyladenosine (m6A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Patil, D.P.; Pickering, B.F.; Jaffrey, S.R. Reading m6A in the Transcriptome: m6A-Binding Proteins. Trends Cell Biol. 2018, 28, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Jaffrey, S.R. A Unified Model for the Function of YTHDF Proteins in Regulating m6A-Modified mRNA. Cell 2020, 181, 1582–1595.e18. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Li, A.; Chen, Y.S.; Ping, X.L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Höfler, S.; Carlomagno, T. Structural and functional roles of 2′-O-ribose methylations and their enzymatic machinery across multiple classes of RNAs. Curr. Opin. Struct. Biol. 2020, 65, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Belanger, F.; Stepinski, J.; Darzynkiewicz, E.; Pelletier, J. Characterization of hMTr1, a human Cap1 2′-O-ribose methyltransferase. J. Biol. Chem. 2010, 285, 33037–33044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature 2019, 565, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Purta, E.; Kaminska, K.H.; Cymerman, I.A.; Campbell, D.A.; Mittra, B.; Zamudio, J.R.; Sturm, N.R.; Jaworski, J.; Bujnicki, J.M. 2′-O-ribose methylation of cap2 in human: Function and evolution in a horizontally mobile family. Nucleic Acids Res. 2011, 39, 4756–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, K.M.; Schaening, C.; Carlile, T.M.; Gilbert, W.V. Conserved Methyltransferase Spb1 Targets mRNAs for Regulated Modification with 2′-O-Methyl Ribose. bioRxiv 2018, 271916. [Google Scholar] [CrossRef]
- Ayadi, L.; Galvanin, A.; Pichot, F.; Marchand, V.; Motorin, Y. RNA ribose methylation (2′-O-methylation): Occurrence, biosynthesis and biological functions. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Oerum, S.; Meynier, V.; Catala, M.; Tisné, C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 2021, 49, 7239–7255. [Google Scholar] [CrossRef]
- Sun, H.; Li, K.; Zhang, X.; Liu, J.; Zhang, M.; Meng, H.; Yi, C. m6Am-seq reveals the dynamic m6Am methylation in the human transcriptome. Nat. Commun. 2021, 12, 4778. [Google Scholar] [CrossRef]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef]
- Boulias, K.; Toczydlowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.J.; Debart, F.; Aravind, L.; et al. Identification of the m6Am Methyltransferase PCIF1 Reveals the Location and Functions of m6Am in the Transcriptome. Mol. Cell 2019, 75, 631–643.e8. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, M.; Xie, D.; Huang, Z.; Zhang, L.; Yang, Y.; Ma, D.; Li, W.; Zhou, Q.; Yang, Y.G.; et al. METTL3-mediated N6-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 2018, 28, 1050–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wnuk, M.; Slipek, P.; Dziedzic, M.; Lewinska, A. The Roles of Host 5-Methylcytosine RNA Methyltransferases during Viral Infections. Int. J. Mol. Sci. 2020, 21, 8176. [Google Scholar] [CrossRef]
- Cristinelli, S.; Angelino, P.; Janowczyk, A.; Delorenzi, M.; Ciuffi, A. HIV Modifies the m6A and m5C Epitranscriptomic Landscape of the Host Cell. Front. Virol. 2021, 1, 714475. [Google Scholar] [CrossRef]
- Guo, G.; Pan, K.; Fang, S.; Ye, L.; Tong, X.; Wang, Z.; Xue, X.; Zhang, H. Advances in mRNA 5-methylcytosine modifications: Detection, effectors, biological functions, and clinical relevance. Mol. Ther. Nucleic Acids 2021, 26, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Wiener, D.; Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2021, 22, 119–131. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.J.; Pan, T.; Kalsotra, A. RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 2017, 18, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, D.G. Post-Transcriptional Regulation of Viral RNA through Epitranscriptional Modification. Cells 2021, 10, 1129. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, S.; Xiao, W.; Zhao, Y.L.; Yang, Y.G. m6A: Signaling for mRNA splicing. RNA Biol. 2016, 13, 756–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Choe, J.; Park, O.H.; Kim, Y.K. Molecular Mechanisms Driving mRNA Degradation by m6A Modification. Trends Genet. 2020, 36, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Yang, Y.; Guo, W.; Xu, L.; You, M.; Zhang, Y.C.; Sun, Z.; Cui, X.; Yu, G.; Qi, Z.; et al. METTL3-dependent m6A modification programs T follicular helper cell differentiation. Nat. Commun. 2021, 12, 1333. [Google Scholar] [CrossRef] [PubMed]
- Fustin, J.M.; Doi, M.; Yamaguchi, Y.; Hida, H.; Nishimura, S.; Yoshida, M.; Isagawa, T.; Morioka, M.S.; Kakeya, H.; Manabe, I.; et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013, 155, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhai, Y.; Zhang, S.; Dai, X.; Li, Z. Roles of N6-Methyladenosine (m6A) in Stem Cell Fate Decisions and Early Embryonic Development in Mammals. Front. Cell Dev. Biol. 2020, 8, 782. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Singh, R.; Shimba, S. Methylated cap structures in eukaryotic RNAs: Structure, synthesis and functions. Pharmacol. Ther. 1992, 54, 249–267. [Google Scholar] [CrossRef]
- Ge, J.; Liu, H.; Yu, Y.T. Regulation of pre-mRNA splicing in Xenopus oocytes by targeted 2′-O-methylation. RNA 2010, 16, 1078–1085. [Google Scholar] [CrossRef] [Green Version]
- Elliott, B.A.; Ho, H.T.; Ranganathan, S.V.; Vangaveti, S.; Ilkayeva, O.; Abou Assi, H.; Choi, A.K.; Agris, P.F.; Holley, C.L. Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat. Commun. 2019, 10, 3401. [Google Scholar] [CrossRef] [Green Version]
- Zust, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Paramasivam, A. RNA 2′-O-methylation modification and its implication in COVID-19 immunity. Cell Death Discov. 2020, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liu, Y.; Rohde, C.; Pauli, C.; Gerloff, D.; Kohn, M.; Misiak, D.; Baumer, N.; Cui, C.; Gollner, S.; et al. AML1-ETO requires enhanced C/D box snoRNA/RNP formation to induce self-renewal and leukaemia. Nat. Cell Biol. 2017, 19, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Jasinski-Bergner, S.; Blumke, J.; Wickenhauser, C.; Seliger, B. Relevance of 2′-O-Methylation and Pseudouridylation for the Malignant Melanoma. Cancers 2021, 13, 1167. [Google Scholar] [CrossRef] [PubMed]
- Tartell, M.A.; Boulias, K.; Hoffmann, G.B.; Bloyet, L.M.; Greer, E.L.; Whelan, S.P.J. Methylation of viral mRNA cap structures by PCIF1 attenuates the antiviral activity of interferon-beta. Proc. Natl. Acad. Sci. USA 2021, 118, e2025769118. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Pestka, S. The Interferons: 50 Years after Their Discovery, There Is Much More to Learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.W.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Hyde, J.L.; Diamond, M.S. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology 2015, 479–480, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.W.; Amarasinghe, G.K. When your cap matters: Structural insights into self vs non-self recognition of 5′ RNA by immunomodulatory host proteins. Curr. Opin. Struct. Biol. 2016, 36, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Zhang, W.; Chen, Y.; Yuan, Q.; Qin, N.N.; Qu, G. The Emerging Role of RNA Modifications in the Regulation of Antiviral Innate Immunity. Front. Microbiol. 2022, 13, 845625. [Google Scholar] [CrossRef]
- Durbin, A.F.; Wang, C.; Marcotrigiano, J.; Gehrke, L. RNAs Containing Modified Nucleotides Fail To Trigger RIG-I Conformational Changes for Innate Immune Signaling. mBio 2016, 7, e00833-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N1-2′O-Methylated Self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, C. Dynamic RNA modifications in posttranscriptional regulation. Mol. Cell 2014, 56, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.G.; Chen, R.; Ahmad, S.; Verma, R.; Kasturi, S.P.; Amaya, L.; Broughton, J.P.; Kim, J.; Cadena, C.; Pulendran, B.; et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 2019, 76, 96–109.e9. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Vasic, R.; Song, Y.; Teng, R.; Liu, C.; Gbyli, R.; Biancon, G.; Nelakanti, R.; Lobben, K.; Kudo, E.; et al. m6A Modification Prevents Formation of Endogenous Double-Stranded RNAs and Deleterious Innate Immune Responses during Hematopoietic Development. Immunity 2020, 52, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Tao, W.; Tong, J.; Gao, J.; Wang, J.; Hou, G.; Qian, C.; Zhang, G.; Li, R.; Wang, D.; et al. m6A modifications regulate intestinal immunity and rotavirus infection. eLife 2022, 11, e73628. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Cai, B.; Zhao, J.; Zhang, L.; Zheng, Y.; Liu, B.; Gao, C. Methyltransferase-Like Protein 14 Attenuates Mitochondrial Antiviral Signaling Protein Expression to Negatively Regulate Antiviral Immunity via N6-methyladenosine Modification. Adv. Sci. 2021, 8, e2100606. [Google Scholar] [CrossRef]
- Chen, J.; Wei, X.; Wang, X.; Liu, T.; Zhao, Y.; Chen, L.; Luo, Y.; Du, H.; Li, Y.; Liu, T.; et al. TBK1-METTL3 axis facilitates antiviral immunity. Cell Rep. 2022, 38, 110373. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Ling, T.; Wang, Y.; Jia, X.; Xie, X.; Chen, R.; Chen, S.; Yuan, S.; Xu, A. Degradation of WTAP blocks antiviral responses by reducing the m6 A levels of IRF3 and IFNAR1 mRNA. EMBO Rep. 2021, 22, e52101. [Google Scholar] [CrossRef] [PubMed]
- McFadden, M.J.; McIntyre, A.B.R.; Mourelatos, H.; Abell, N.S.; Gokhale, N.S.; Ipas, H.; Xhemalçe, B.; Mason, C.E.; Horner, S.M. Post-transcriptional regulation of antiviral gene expression by N6-methyladenosine. Cell Rep. 2021, 34, 108798. [Google Scholar] [CrossRef]
- Shaw, A.E.; Hughes, J.; Gu, Q.; Behdenna, A.; Singer, J.B.; Dennis, T.; Orton, R.J.; Varela, M.; Gifford, R.J.; Wilson, S.J.; et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 2017, 15, e2004086. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Rana, T.M. Regulation of antiviral innate immunity by chemical modification of viral RNA. Wiley Interdiscip. Rev. RNA 2022, e1720. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Hui, H.; Bray, B.; Gonzalez, G.M.; Zeller, M.; Anderson, K.G.; Knight, R.; Smith, D.; Wang, Y.; Carlin, A.F.; et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 2021, 35, 109091. [Google Scholar] [CrossRef]
- Kastan, J.P.; Tremblay, M.W.; Brown, M.C.; Trimarco, J.D.; Dobrikova, E.Y.; Dobrikov, M.I.; Gromeier, M. Enterovirus 2Apro Cleavage of the YTHDF m6A Readers Implicates YTHDF3 as a Mediator of Type I Interferon-Driven JAK/STAT Signaling. mBio 2021, 12, e00116-21. [Google Scholar] [CrossRef]
- Gokhale, N.S.; McIntyre, A.B.R.; Mattocks, M.D.; Holley, C.L.; Lazear, H.M.; Mason, C.E.; Horner, S.M. Altered m6A Modification of Specific Cellular Transcripts Affects Flaviviridae Infection. Mol. Cell 2020, 77, 542–555.e548. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Imam, H.; Khan, M.; Mir, S.A.; Kim, S.J.; Yoon, S.K.; Hur, W.; Siddiqui, A. HBV-Induced Increased N6 Methyladenosine Modification of PTEN RNA Affects Innate Immunity and Contributes to HCC. Hepatology 2021, 73, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Imam, H.; Khan, M.; Siddiqui, A. N 6-Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J. Biol. Chem. 2020, 295, 13123–13133. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Zhang, Q.; Zhang, R.; Lu, Y.; Wang, X.; Tian, H.; Yang, Y.; Gu, Z.; Gao, Y.; Yang, X.; et al. N6-methyladenosine RNA modification suppresses antiviral innate sensing pathways via reshaping double-stranded RNA. Nat. Commun. 2021, 12, 1582. [Google Scholar] [CrossRef]
- Lu, M.; Xue, M.; Wang, H.T.; Kairis, E.L.; Ahmad, S.; Wei, J.; Zhang, Z.; Liu, Q.; Zhang, Y.; Gao, Y.; et al. Nonsegmented Negative-Sense RNA Viruses Utilize N 6-Methyladenosine (m6A) as a Common Strategy To Evade Host Innate Immunity. J. Virol. 2021, 95, e01939-20. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Z.; Xue, M.; Zhao, B.S.; Harder, O.; Li, A.; Liang, X.; Gao, T.Z.; Xu, Y.; Zhou, J.; et al. N6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 2020, 5, 584–598. [Google Scholar] [CrossRef]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cai, Y.; Ma, Z.; Jiang, B.; Liu, W.; Cheng, J.; Guo, N.; Wang, Z.; Sealy, J.E.; Song, C.; et al. The RNA helicase DDX5 promotes viral infection via regulating N6-methyladenosine levels on the DHX58 and NFkappaB transcripts to dampen antiviral innate immunity. PLoS Pathog. 2021, 17, e1009530. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Li, Z.; Cao, X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 2017, 18, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kumar, S.; Espada, C.E.; Tirumuru, N.; Cahill, M.P.; Hu, L.; He, C.; Wu, L. N6-methyladenosine modification of HIV-1 RNA suppresses type-I interferon induction in differentiated monocytic cells and primary macrophages. PLoS Pathog. 2021, 17, e1009421. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Siddiqui, A. Hepatitis B virus X protein recruits methyltransferases to affect cotranscriptional N6-methyladenosine modification of viral/host RNAs. Proc. Natl. Acad. Sci. USA 2021, 118, e2019455118. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Kim, G.W.; Mir, S.A.; Khan, M.; Siddiqui, A. Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified Hepatitis B Virus transcripts. PLoS Pathog. 2020, 16, e1008338. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Siddiqui, A. Hepatitis B Virus X Protein Expression Is Tightly Regulated by N6-Methyladenosine Modification of Its mRNA. J. Virol. 2022, 96, e0165521. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.M.; Depledge, D.P.; Bianco, C.; Thompson, L.; Mohr, I. RNA m6 A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 2018, 32, 1472–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, P. The dsRNA viruses. Virus Res. 2004, 101, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Rampersad, S.; Tennant, P. Replication and Expression Strategies of Viruses. Viruses 2018, 55–82. [Google Scholar] [CrossRef]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Primers 2017, 3, 17083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, S.M.; Patton, J.T. Assortment and packaging of the segmented rotavirus genome. Trends Microbiol. 2011, 19, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Uzri, D.; Greenberg, H.B. Characterization of rotavirus RNAs that activate innate immune signaling through the RIG-I-like receptors. PLoS ONE 2013, 8, e69825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Luongo, C.L.; Nibert, M.L.; Patton, J.T. Rotavirus open cores catalyze 5′-capping and methylation of exogenous RNA: Evidence that VP3 is a methyltransferase. Virology 1999, 265, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Yu, X.; Crawford, S.E.; Moreno, R.; Jakana, J.; Sankaran, B.; Anish, R.; Kaundal, S.; Hu, L.; Estes, M.K.; et al. 2.7 A cryo-EM structure of rotavirus core protein VP3, a unique capping machine with a helicase activity. Sci. Adv. 2020, 6, eaay6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, M.; Ogden, K.M.; Patton, J.T. Silencing the alarms: Innate immune antagonism by rotavirus NSP1 and VP3. Virology 2015, 479–480, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Zhu, S.; Ren, L.; Feng, N.; Song, Y.; Ge, X.; Li, B.; Flavell, R.A.; Greenberg, H.B. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. eLife 2018, 7, e39494. [Google Scholar] [CrossRef]
- Arnold, M.M.; Brownback, C.S.; Taraporewala, Z.F.; Patton, J.T. Rotavirus variant replicates efficiently although encoding an aberrant NSP3 that fails to induce nuclear localization of poly(A)-binding protein. J. Gen. Virol. 2012, 93, 1483–1494. [Google Scholar] [CrossRef] [Green Version]
- Gratia, M.; Sarot, E.; Vende, P.; Charpilienne, A.; Baron, C.H.; Duarte, M.; Pyronnet, S.; Poncet, D. Rotavirus NSP3 Is a Translational Surrogate of the Poly(A) Binding Protein-Poly(A) Complex. J. Virol. 2015, 89, 8773–8782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaconis, G.; Jackson, B.; Childs, K.; Boyce, M.; Goodbourn, S.; Blake, N.; Iturriza-Gomara, M.; Seago, J. Rotavirus NSP1 Inhibits Type I and Type III Interferon Induction. Viruses 2021, 13, 589. [Google Scholar] [CrossRef] [PubMed]
- Apte-Sengupta, S.; Sirohi, D.; Kuhn, R.J. Coupling of replication and assembly in flaviviruses. Curr. Opin. Virol. 2014, 9, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Comas-Garcia, M. Packaging of Genomic RNA in Positive-Sense Single-Stranded RNA Viruses: A Complex Story. Viruses 2019, 11, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichinchi, G.; Zhao, B.S.; Wu, Y.; Lu, Z.; Qin, Y.; He, C.; Rana, T.M. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe 2016, 20, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Dong, H.; Chen, H.; Zhang, J.; Ling, H.; Li, Z.; Shi, P.Y.; Li, H. Flavivirus RNA cap methyltransferase: Structure, function, and inhibition. Front. Biol. 2010, 5, 286–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Soh, T.S.; Lim, S.P.; Chung, K.Y.; Swaminathan, K.; Vasudevan, S.G.; Shi, P.Y.; Lescar, J.; Luo, D. Molecular basis for specific viral RNA recognition and 2′-O-ribose methylation by the dengue virus nonstructural protein 5 (NS5). Proc. Natl. Acad. Sci. USA 2015, 112, 14834–14839. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.C.; Hoang, L.T.; Mohamed Naim, A.N.; Dong, H.; Schreiber, M.J.; Hibberd, M.L.; Tan, M.J.A.; Shi, P.Y. Evasion of early innate immune response by 2′-O-methylation of dengue genomic RNA. Virology 2016, 499, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.Y.; Li, H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M., Jr.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.H.; Dong, H.; Li, X.F.; Xie, X.; Zhao, H.; Deng, Y.Q.; Wang, X.Y.; Ye, Q.; Zhu, S.Y.; Wang, H.J.; et al. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J. Virol. 2013, 87, 5812–5819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zust, R.; Dong, H.; Li, X.F.; Chang, D.C.; Zhang, B.; Balakrishnan, T.; Toh, Y.X.; Jiang, T.; Li, S.H.; Deng, Y.Q.; et al. Rational design of a live attenuated dengue vaccine: 2′-o-methyltransferase mutants are highly attenuated and immunogenic in mice and macaques. PLoS Pathog. 2013, 9, e1003521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, S.M. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signaling. J. Virol. 2017, 91, e01970-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wu, Z.; Wang, M.; Cheng, A. Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins. Viruses 2017, 9, 291. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Chang, D.C.; Hua, M.H.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.M.; Dedon, P.C.; et al. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 2012, 8, e1002642. [Google Scholar] [CrossRef]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.Y.; Chen, S.; Zhao, X.; Lei, C.Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Roth, H.; Magg, V.; Uch, F.; Mutz, P.; Klein, P.; Haneke, K.; Lohmann, V.; Bartenschlager, R.; Fackler, O.T.; Locker, N.; et al. Flavivirus Infection Uncouples Translation Suppression from Cellular Stress Responses. mBio 2017, 8, e02150-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, K.; Oshiumi, H.; Takaki, H.; Matsumoto, M.; Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015, 11, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Tang, K.; Chen, D.; Hong, M.; Sun, F.; Wang, S.; Ke, Y.; Wu, T.; Sun, R.; Qian, J.; et al. Riok3 inhibits the antiviral immune response by facilitating TRIM40-mediated RIG-I and MDA5 degradation. Cell Rep. 2021, 35, 109272. [Google Scholar] [CrossRef]
- Feng, J.; De Jesus, P.D.; Su, V.; Han, S.; Gong, D.; Wu, N.C.; Tian, Y.; Li, X.; Wu, T.T.; Chanda, S.K.; et al. RIOK3 is an adaptor protein required for IRF3-mediated antiviral type I interferon production. J. Virol. 2014, 88, 7987–7997. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Imbert, I.; Coutard, B.; Bouvet, M.; Selisko, B.; Alvarez, K.; Gorbalenya, A.E.; Snijder, E.J.; Canard, B. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J. Virol. 2008, 82, 8071–8084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Su, C.; Ke, M.; Jin, X.; Xu, L.; Zhang, Z.; Wu, A.; Sun, Y.; Yang, Z.; Tien, P.; et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011, 7, e1002294. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Yount, B.L., Jr.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-o-methyltransferase activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menachery, V.D.; Gralinski, L.E.; Mitchell, H.D.; Dinnon, K.H., 3rd; Leist, S.R.; Yount, B.L., Jr.; Graham, R.L.; McAnarney, E.T.; Stratton, K.G.; Cockrell, A.S.; et al. Middle East Respiratory Syndrome Coronavirus Nonstructural Protein 16 Is Necessary for Interferon Resistance and Viral Pathogenesis. mSphere 2017, 2, e00346-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.M.; Depledge, D.P.; Thompson, L.; Srinivas, K.P.; Grande, R.C.; Vink, E.I.; Abebe, J.S.; Blackaby, W.P.; Hendrick, A.; Albertella, M.R.; et al. Targeting the m6A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev. 2021, 35, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, Y.P.; Li, K.; Ye, Q.; Zhou, H.Y.; Sun, H.; Li, X.; Yu, L.; Deng, Y.Q.; Li, R.T.; et al. The m6A methylome of SARS-CoV-2 in host cells. Cell Res. 2021, 31, 404–414. [Google Scholar] [CrossRef]
- Zhang, X.; Hao, H.; Ma, L.; Zhang, Y.; Hu, X.; Chen, Z.; Liu, D.; Yuan, J.; Hu, Z.; Guan, W. Methyltransferase-like 3 Modulates Severe Acute Respiratory Syndrome Coronavirus-2 RNA N6-Methyladenosine Modification and Replication. mBio 2021, 12, e0106721. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.W.; Wu, X.X.; Jiang, X.G.; Xu, K.J.; Ying, L.J.; Ma, C.L.; Li, S.B.; Wang, H.Y.; Zhang, S.; Gao, H.N.; et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: Retrospective case series. BMJ 2020, 368, m606. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.R.; Sarnow, P. Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology 2003, 315, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Van der Linden, L.; Wolthers, K.C.; van Kuppeveld, F.J. Replication and Inhibitors of Enteroviruses and Parechoviruses. Viruses 2015, 7, 4529–4562. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytonen, V.P.; Flodstrom-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J. Virol. 2016, 90, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Correction: Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS Pathog. 2017, 13, e1006243. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Hao, S.; Chen, H.; Chen, Z.; Zhang, Y.; Wang, J.; Wang, H.; Zhang, B.; Qiu, J.; Deng, F.; et al. N6-methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res. 2019, 47, 362–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, M.; Dong, Y.; Wang, Y.; Liu, H.; Ma, H.; Zhang, H.; Zhang, L.; Cheng, L.; Lv, X.; Xu, Z.; et al. N6-methyladenosine modifications enhance enterovirus 71 ORF translation through METTL3 cytoplasmic distribution. Biochem. Biophys. Res. Commun. 2020, 527, 297–304. [Google Scholar] [CrossRef]
- Xue, M.; Zhang, Y.; Wang, H.; Kairis, E.L.; Lu, M.; Ahmad, S.; Attia, Z.; Harder, O.; Zhang, Z.; Wei, J.; et al. Viral RNA N6-methyladenosine modification modulates both innate and adaptive immune responses of human respiratory syncytial virus. PLoS Pathog. 2021, 17, e1010142. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Green, T.J. RNA Synthesis and Capping by Non-segmented Negative Strand RNA Viral Polymerases: Lessons From a Prototypic Virus. Front. Microbiol. 2019, 10, 1490. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.T.; Whelan, S.P. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2006, 103, 8493–8498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011, 10, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crepin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Longhi, S.; Henrissat, B.; Canard, B. Viral RNA-polymerases—A predicted 2′-O-ribose methyltransferase domain shared by all Mononegavirales. Trends Biochem. Sci. 2002, 27, 222–224. [Google Scholar] [CrossRef]
- Ogino, T. Capping of vesicular stomatitis virus pre-mRNA is required for accurate selection of transcription stop-start sites and virus propagation. Nucleic Acids Res. 2014, 42, 12112–12125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paesen, G.C.; Collet, A.; Sallamand, C.; Debart, F.; Vasseur, J.J.; Canard, B.; Decroly, E.; Grimes, J.M. X-ray structure and activities of an essential Mononegavirales L-protein domain. Nat. Commun. 2015, 6, 8749. [Google Scholar] [CrossRef]
- Li, J.; Fontaine-Rodriguez, E.C.; Whelan, S.P. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J. Virol. 2005, 79, 13373–13384. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wei, Y.; Zhang, X.; Zhang, Y.; Cai, H.; Zhu, Y.; Shilo, K.; Oglesbee, M.; Krakowka, S.; Whelan, S.P.; et al. mRNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J. Virol. 2014, 88, 2913–2926. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Zhao, B.S.; Zhang, Z.; Lu, M.; Harder, O.; Chen, P.; Lu, Z.; Li, A.; Ma, Y.; Xu, Y.; et al. Viral N6-methyladenosine upregulates replication and pathogenesis of human respiratory syncytial virus. Nat. Commun. 2019, 10, 4595. [Google Scholar] [CrossRef]
- Courtney, D.G.; Kennedy, E.M.; Dumm, R.E.; Bogerd, H.P.; Tsai, K.; Heaton, N.S.; Cullen, B.R. Epitranscriptomic Enhancement of Influenza A Virus Gene Expression and Replication. Cell Host Microbe 2017, 22, 377–386.e5. [Google Scholar] [CrossRef] [Green Version]
- Thammawat, S.; Sadlon, T.A.; Hallsworth, P.G.; Gordon, D.L. Role of cellular glycosaminoglycans and charged regions of viral G protein in human metapneumovirus infection. J. Virol. 2008, 82, 11767–11774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Strebel, K. HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 2013, 3, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramdas, P.; Sahu, A.K.; Mishra, T.; Bhardwaj, V.; Chande, A. From Entry to Egress: Strategic Exploitation of the Cellular Processes by HIV-1. Front. Microbiol. 2020, 11, 559792. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J. Putting an ‘End’ to HIV mRNAs: Capping and polyadenylation as potential therapeutic targets. AIDS Res. Ther. 2013, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.L.; Ho, C.K.; Saha, N.; Schwer, B.; Shuman, S.; Rana, T.M. Tat stimulates cotranscriptional capping of HIV mRNA. Mol. Cell 2002, 10, 585–597. [Google Scholar] [CrossRef]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G.M.; Bansal, V.; Wang, Y.; Mason, C.E.; Rana, T.M. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016, 1, 16011. [Google Scholar] [CrossRef] [PubMed]
- Tirumuru, N.; Zhao, B.S.; Lu, W.; Lu, Z.; He, C.; Wu, L. N6-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife 2016, 5, e15528. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Bogerd, H.P.; Kornepati, A.V.R.; Kang, D.; Ghoshal, D.; Marshall, J.B.; Poling, B.C.; Tsai, K.; Gokhale, N.S.; Horner, S.M.; et al. Posttranscriptional m6A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe 2017, 22, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, D.G.; Tsai, K.; Bogerd, H.P.; Kennedy, E.M.; Law, B.A.; Emery, A.; Swanstrom, R.; Holley, C.L.; Cullen, B.R. Epitranscriptomic Addition of m5C to HIV-1 Transcripts Regulates Viral Gene Expression. Cell Host Microbe 2019, 26, 217–227.e6. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Bogerd, H.P.; Kennedy, E.M.; Emery, A.; Swanstrom, R.; Cullen, B.R. Epitranscriptomic addition of m6A regulates HIV-1 RNA stability and alternative splicing. Genes Dev. 2021, 35, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, K.; Song, B.; Ma, J.; Wu, X.; Xu, Q.; Wei, Z.; Su, J.; Liu, G.; Rong, R.; et al. m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021, 49, D134–D143. [Google Scholar] [CrossRef] [PubMed]
- Selberg, S.; Zusinaite, E.; Herodes, K.; Seli, N.; Kankuri, E.; Merits, A.; Karelson, M. HIV Replication Is Increased by RNA Methylation METTL3/METTL14/WTAP Complex Activators. ACS Omega 2021, 6, 15957–15963. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Tirumuru, N.; St Gelais, C.; Koneru, P.C.; Liu, C.; Kvaratskhelia, M.; He, C.; Wu, L. N6-Methyladenosine-binding proteins suppress HIV-1 infectivity and viral production. J. Biol. Chem. 2018, 293, 12992–13005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, A.; Caroe, E.R.; Barr, G.C.R.; Bishop, K.N. The Role of Capsid in HIV-1 Nuclear Entry. Viruses 2021, 13, 1425. [Google Scholar] [CrossRef] [PubMed]
- Zila, V.; Margiotta, E.; Turonova, B.; Muller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Borner, K.; Rada, J.; Muller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef] [PubMed]
- Yedavalli, V.S.; Jeang, K.T. Trimethylguanosine capping selectively promotes expression of Rev-dependent HIV-1 RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 14787–14792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Kang, Y.; Wang, S.; Gonzalez, G.M.; Li, W.; Hui, H.; Wang, Y.; Rana, T.M. HIV reprograms host m6Am RNA methylome by viral Vpr protein-mediated degradation of PCIF1. Nat. Commun. 2021, 12, 5543. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, H.; Lian, Z.; Deng, S. The Role of RNA Modification in HIV-1 Infection. Int. J. Mol. Sci. 2022, 23, 7571. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [Green Version]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Kim, G.W.; Siddiqui, A. Epitranscriptomic(N6-methyladenosine) Modification of Viral RNA and Virus-Host Interactions. Front. Cell. Infect. Microbiol. 2020, 10, 584283. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Nie, H.; Mao, R.; Mitra, B.; Cai, D.; Yan, R.; Guo, J.T.; Block, T.M.; Mechti, N.; Guo, H. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog. 2017, 13, e1006296. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Khan, M.; Gokhale, N.S.; McIntyre, A.B.R.; Kim, G.W.; Jang, J.Y.; Kim, S.J.; Mason, C.E.; Horner, S.M.; Siddiqui, A. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl. Acad. Sci. USA 2018, 115, 8829–8834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espert, L.; Degols, G.; Lin, Y.L.; Vincent, T.; Benkirane, M.; Mechti, N. Interferon-induced exonuclease ISG20 exhibits an antiviral activity against human immunodeficiency virus type 1. J. Gen. Virol. 2005, 86, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.Y.; et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, N.; Woodson, S.E.; Dong, Q.; Wang, J.; Liang, Y.; Rijnbrand, R.; Wei, L.; Nichols, J.E.; Guo, J.T.; et al. Antiviral activities of ISG20 in positive-strand RNA virus infections. Virology 2011, 409, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsano, C.; Billet, O.; Bennoun, M.; Cavard, C.; Zider, A.; Grimber, G.; Natoli, G.; Briand, P.; Levrero, M. Hepatitis B virus X gene product acts as a transactivator in vivo. J. Hepatol. 1994, 21, 103–109. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Shen, F.; Wang, Y.; Li, Z.; Chen, J.; Yuan, Z. Residues Asn118 and Glu119 of hepatitis B virus X protein are critical for HBx-mediated inhibition of RIG-I-MAVS signaling. Virology 2020, 539, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, D.; Krupovic, M.; Venclovas, C. The logic of DNA replication in double-stranded DNA viruses: Insights from global analysis of viral genomes. Nucleic Acids Res. 2016, 44, 4551–4564. [Google Scholar] [CrossRef] [Green Version]
- Moss, B.; Gershowitz, A.; Stringer, J.R.; Holland, L.E.; Wagner, E.K. 5′-Terminal and internal methylated nucleosides in herpes simplex virus type 1 mRNA. J. Virol. 1977, 23, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnierle, B.S.; Gershon, P.D.; Moss, B. Cap-specific mRNA (nucleoside-O2′-)-methyltransferase and poly(A) polymerase stimulatory activities of vaccinia virus are mediated by a single protein. Proc. Natl. Acad. Sci. USA 1992, 89, 2897–2901. [Google Scholar] [CrossRef] [Green Version]
- De la Pena, M.; Kyrieleis, O.J.; Cusack, S. Structural insights into the mechanism and evolution of the vaccinia virus mRNA cap N7 methyl-transferase. EMBO J. 2007, 26, 4913–4925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.; Liu, H.; Zhang, S.; da Silva, S.R.; Zhang, L.; Meng, J.; Cui, X.; Yuan, H.; Sorel, O.; Zhang, S.W.; et al. Viral and cellular N6-methyladenosine and N6,2′-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 2018, 3, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Courtney, D.G.; Cullen, B.R. Addition of m6A to SV40 late mRNAs enhances viral structural gene expression and replication. PLoS Pathog. 2018, 14, e1006919. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Hayer, K.E.; McIntyre, A.B.R.; Gokhale, N.S.; Abebe, J.S.; Della Fera, A.N.; Mason, C.E.; Horner, S.M.; Wilson, A.C.; Depledge, D.P.; et al. Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat. Commun. 2020, 11, 6016. [Google Scholar] [CrossRef]
- Altman, A.M.; Mahmud, J.; Nikolovska-Coleska, Z.; Chan, G. HCMV modulation of cellular PI3K/AKT/mTOR signaling: New opportunities for therapeutic intervention? Antivir. Res. 2019, 163, 82–90. [Google Scholar] [CrossRef]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, C.; Perez, C.; Mohr, I. Poly(A) binding protein abundance regulates eukaryotic translation initiation factor 4F assembly in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2012, 109, 5627–5632. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Xuan, B.; Liu, H.; Zhong, J.; Yu, D.; Qian, Z. Tuberous Sclerosis Complex Protein 2-Independent Activation of mTORC1 by Human Cytomegalovirus pUL38. J. Virol. 2015, 89, 7625–7635. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class III: dsRNA Viruses | ||||||||
| Viral Family | Virus Name | Chemical Modification/ Machinery | Analyzed Modified RNA | Detection Technique | Main Outcome(s) | Impact on Replication | Impact on Host Immune Response | Ref. |
| Reoviridae | Rotavirus (RV) | m6A | Viral genome and host IRF7 mRNA | m6A-seq1, m6A-qPCR2. | -Presence of m6A methylation on viral mRNAs, higher prevalence on NSP3 mRNAs -Viral infection increases m6A methylation levels of host transcripts, by inhibiting ALKBH5 protein expression via viral NSP1 protein, including IRF7, leading to lower mRNA stability, thereby impairing IFN-I signaling Note: In vivo validation in murine model | n.a. | (-) Decreased IRF7 mRNA stability and subsequent IFN-I signaling | [93] |
| Class IV: ssRNA (+) Viruses | ||||||||
| Viral Family | Virus Name | Chemical Modification/ Machinery | Analyzed Modified RNA | Detection Technique | Main Outcome(s) | Impact on Replication | Impact on Host Immune Response | Ref. |
| Coronaviridae | Severe Acute Respiratory Syndrome Coranavirus 2 (SARS-CoV-2) | m6A, m6,6A, 2′-O, ac4C, m3C, m5C, Ψ, m5U | Viral genome | LC-MS/MS-MS/MS/MS3 and m6A-seq | -Viral genome is highly methylated and the presence of m6A modifications on viral RNA impair RIG-I binding and consequently inhibit IFN-I signaling cascade -M6A methylations of host transcripts upon infection inhibit the expression of pro-inflammatory cytokines Note: Severe COVID-19 patients correlate with a lower expression of m6A writers and higher induction of inflammatory genes | + | (-) Decreased RIG-I sensing | [101] |
| Picornaviridae | Enterovirus (EV) | m6A machinery | n.a. | n.a. | -Enterovirus protease 2A cleaves YTHDF1-3 early in the infection phase, leading to suppression of the JAK/STAT signaling pathway | + | (-) Decreased JAK/STAT signaling | [102] |
| Flaviviridae | Dengue Virus (DENV) | m6A | Host RIOK3 mRNA | m6A-seq | -Viral infection modulates m6A methylations of host transcripts involved in infection regulation, including RIOK3 with increased m6A levels leading to induce translation and affecting IFN-I signaling | + | (-) Phosphorylation and inactivation of MDA5 sensor | [103] |
| Flaviviridae | Zika Virus (ZIKV) | m6A | Host RIOK3 mRNA | m6A-seq | -Viral infection modulates m6A methylations of host transcripts involved in infection regulation, including RIOK3 with increased m6A levels leading to induced translation and affecting IFN-I signaling | + | (-) Phosphorylation and inactivation of MDA5 sensor | [103] |
| Hepatitis C Virus (HCV) | m6A | Host RIOK3 mRNA | m6A-seq | -Viral infection modulates m6A methylations of host transcripts involved in infection regulation, including RIOK3 with increased m6A levels leading to induced translation and affecting IFN-I signaling | - | (+) Increased TBK1-IRF3 interaction leading to increased IFN signaling | [103] | |
| m6A | Host PTEN mRNA | m6A-seq | -Virally induced m6A methylation of PTEN results in mRNA degradation via YTHDF2 binding, leading to cytoplasmic retention of IRF3 and inhibited IFN-I signaling pathway | + | (-) Decreased IRF3 nuclear import and subsequent IFN-I signaling | [104] | ||
| m6A | Viral genome | n.a. | -Presence of m6A modification on viral RNA results in an impaired RIG-I sensing and a decreased IFN-I response via YTHDF2 binding | + | (-) Decreased RIG-I sensing | [105] | ||
| m6A machinery | n.a. | n.a. | -METTL3 acts as a negative regulator of the IFNβ innate immunity cascade in response to infection | n.a. | (-) Decreased IFNβ signaling | [106] | ||
| Class V: ssRNA (−) Viruses | ||||||||
| Viral Family | Virus Name | Chemical Modification/ Machinery | Analyzed Modified RNA | Detection Technique | Main Outcome(s) | Impact on Replication | Impact on Host Immune Response | Ref. |
| Paramyxoviridae | Sendai Virus (SeV) | m6A machinery | n.a. | n.a. | -METTL3 translocates to the cytoplasm and negatively regulates IFNβ innate immunity cascade in response to infection | n.a. | (-) Decreased IFN-I signaling | [106] |
| m6A | Viral genome, antigenome and transcripts | m6A-seq | -Presence of m6A on viral RNAs impairs RIG-I activation and hinders IFN-I response | n.a. | (-) Decreased RIG-I sensing | [107] | ||
| Pneumoviridae | Human Metapneumovirus (HMPV) | m6A | Viral genome, antigenome and transcripts | m6A-seq | -m6A methylation of viral RNAs impairs RIG-I binding and the conformational change necessary to activate sensing and IFN-I response Note: In vivo validation in murine model provided | + | (-) Decreased RIG-I sensing | [108] |
| m6A | Viral genome, antigenome and transcripts | m6A-seq | -Presence of m6A on viral RNAs impairs RIG-I activation and hinders IFN-I response | n.a. | (-) Decreased RIG-I sensing | [107] | ||
| Human Respiratory Syncytial Virus (RSV) | m6A | Viral genome, antigenome and transcripts | m6A-seq | -Presence of m6A on viral RNAs impairs RIG-I activation and hinders IFN-I response | n.a. | (-) Decreased RIG-I sensing | [107] | |
| Orthomyxoviridae | Influenza A virus (IAV) | m6A | Host IFNB mRNA | n.a. | -Viral infection induces m6A methylation of IFNB mRNA, leading to transcript destabilization and subsequent impairment of signaling cascade | + | (-) Decreased IFN-I signaling | [109] |
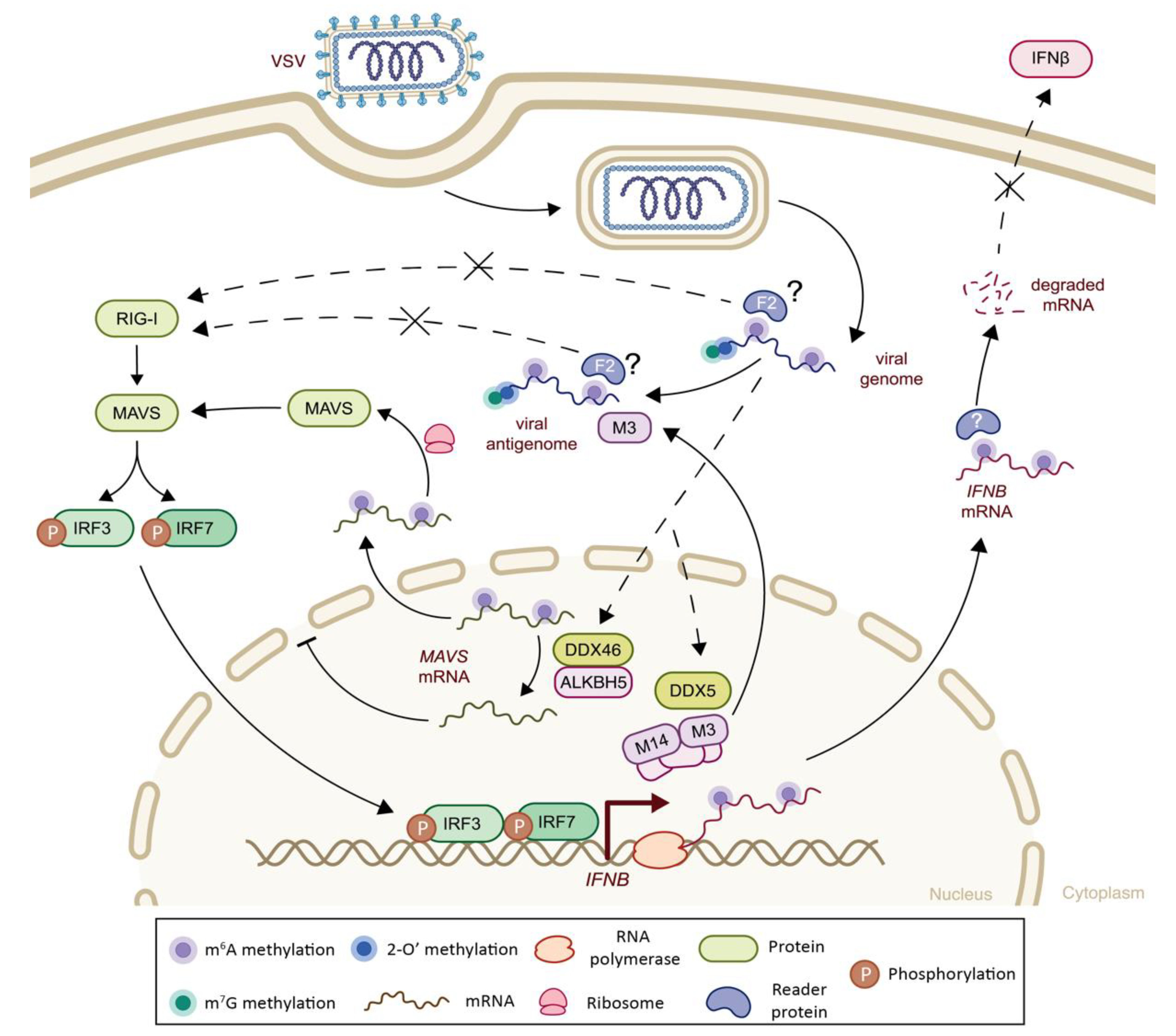
| Rhabdoviridae | Vesicular Stomatitis Virus (VSV) | m6A | Viral genome, antigenome and transcripts | m6A-seq | -Presence of m6A on viral RNAs impairs RIG-I activation and hinders IFN-I response | n.a. | (-) Decreased RIG-I sensing | [107] |
| m6A | Host IFNB mRNA | n.a. | -m6A methylations of IFNB mRNA leading to transcript destabilization and subsequent impairment of signaling cascade | + | (-) Decreased IFN-I signaling | [109] | ||
| m6A | Viral antigenome and transcripts | miCLIP-seq4 and m6A-qPCR | -METTL3 translocates to the cytoplasm and promotes m6A modification on viral transcripts in response to infection, and negatively regulates IFNβ innate immunity cascade -Increase in m6A modifications reduces formation of viral dsRNA, thereby attenuating RLR sensing and IFN-I signaling cascade | + | (-) Decreased RLR sensing, decreased IFNβ signaling | [106] | ||
| Rhabdoviridae | Vesicular Stomatitis Virus (VSV) | m6A | Host p65 and IKKγ mRNAs | m6A-qPCR | -Upon infection, DDX5 interacts with the METTL3–METTL14 complex promoting m6A modification of p65 and IKKγ and their consequent degradation by YTHDF2, resulting in suppression of innate immune response Note: In vivo validation in murine model provided | + | (-) Decreased NF-κβ signaling pathway | [110] |
| m6A | Host TRAF3, TRAF6 and MAVS mRNAs | m6A-qPCR | -Upon infection, DDX46 recruits ALKBH5 and induces demethylation of transcripts involved in antiviral signaling (TRAF3, TRAF6 and MAVS), resulting in their nuclear retention, impaired translation and inhibition of IFN-I signaling | + | (-) Decreased IFN-I signaling | [111] | ||
| m6Am | Viral transcripts | 2D-TLC5 | -The first transcribed adenosine of viral mRNAs is m6Am-methylated, leading to impaired IFN-I response | n.e. | (-) Decreased IFN-I signaling | [73] | ||
| Class VI: ssRNA (+) RT Viruses | ||||||||
| Viral Family | Virus Name | Chemical Modification/ Machinery | Analyzed Modified RNA | Detection Technique | Main Outcome(s) | Impact on Replication | Impact on Host Immune Response | Ref. |
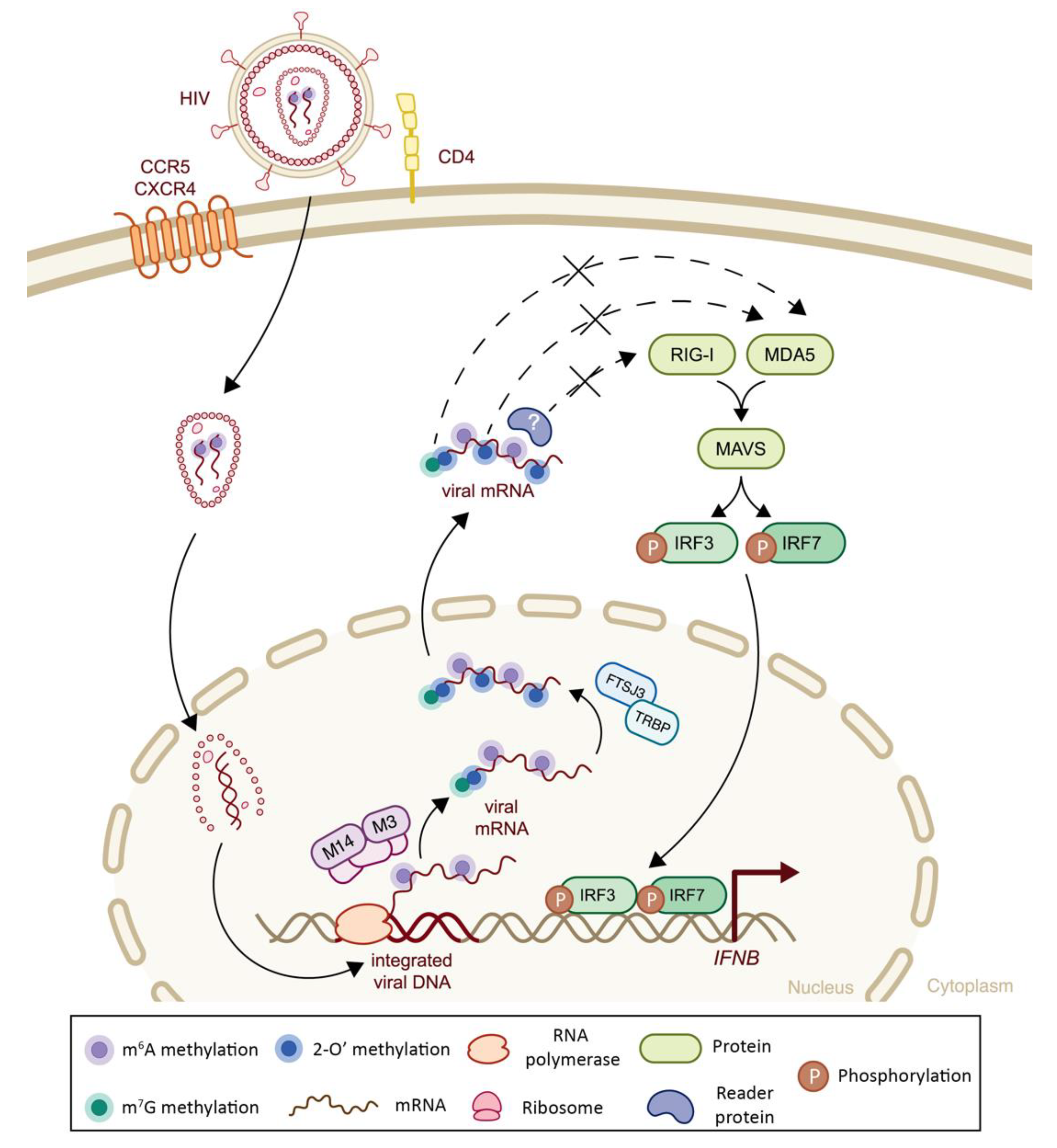
| Retroviridae | Human Immunodeficiency Virus (HIV) | 2′-O | Viral genome | RiboMethSeq6 | -Recruitment of FTSJ3 by TRBP to viral RNA leads to catalyzation of internal 2′O ribose methylations, which impair MDA5 sensing and IFN-I signaling cascade | + | (-) Decreased MDA5 sensing | [43] |
| m6A | Viral transcripts | n.a. | -m6A methylation of viral RNA impairs RIG-I sensing and consequent IFN-I response | + | (-) Decreased RIG-I sensing | [112] | ||
| Class III: dsRNA viruses | ||||||||
| Viral Family | Virus Name | Chemical Modification/ Machinery | Analyzed Modified RNA | Detection Technique | Main Outcome(s) | Impact on Replication | Impact on Host Immune Response | Ref. |
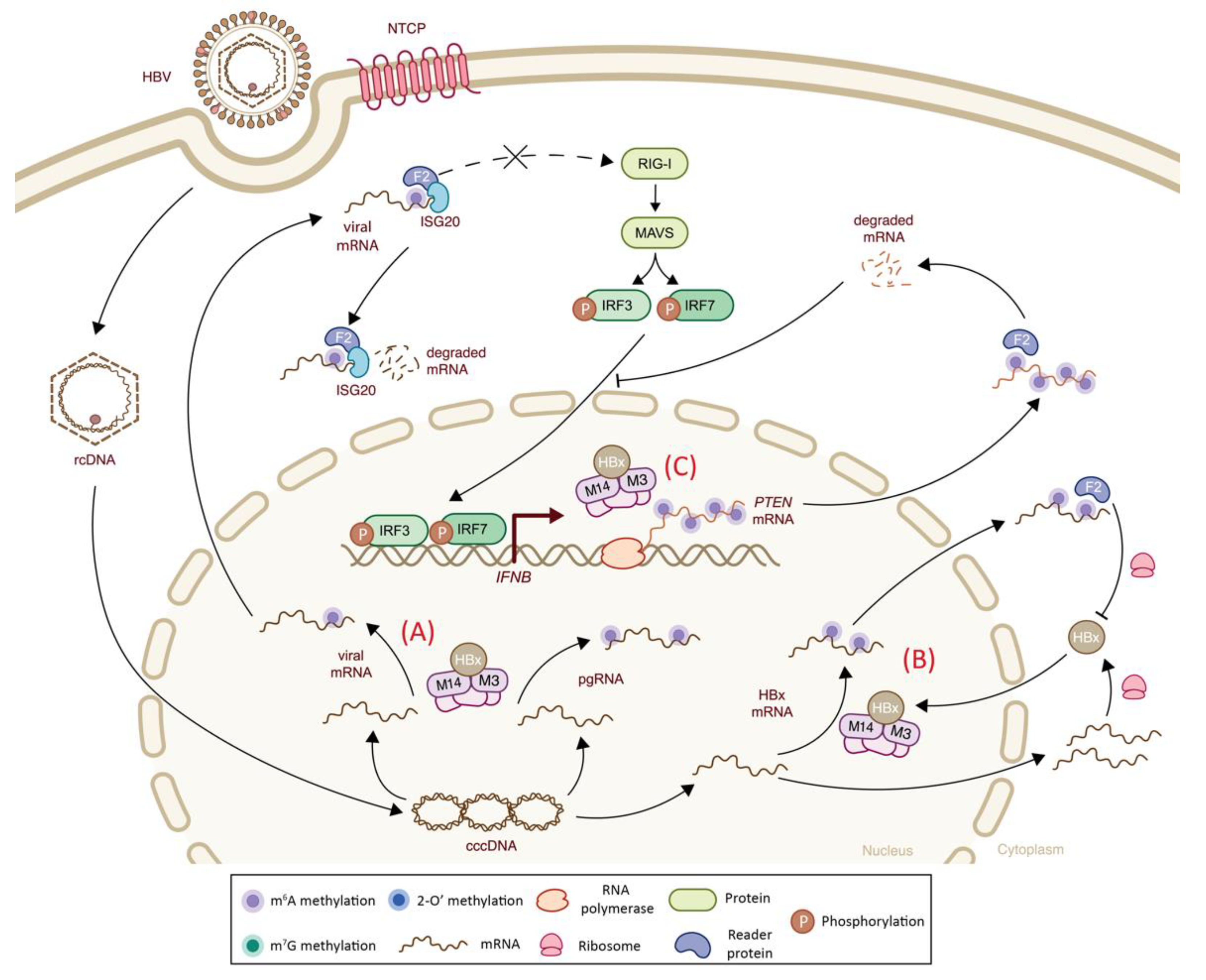
| Hepadnaviridae | Hepatitis B virus (HBV) | m6A | Viral transcripts | n.a. | -Presence of m6A modification on viral RNA results in an impaired RIG-I sensing and a decreased IFN-I response via YTHDF2 binding | + | (-) Decreased RIG-I sensing | [105] |
| Hepadnaviridae | Hepatitis B virus (HBV) | m6A | Host PTEN mRNA | m6A-seq | -Virally induced m6A methylation of PTEN results in mRNA degradation via YTHDF2 binding, leading to cytoplasmic retention of IRF3 and inhibited IFN-I signaling pathway | + | (-) Decreased IRF-3 nuclear import and subsequent IFNβ signaling | [104] |
| m6A | Viral transcript and host PTEN transcript | m6A-qPCR | -Viral HBx protein recruits METTL3–METTL14 complex to catalyze m6A methylation of viral mRNAs and increases host PTEN mRNA methylation that alters IFN-I response | n.a. | (-) Indirect effect on IFN-I signaling | [113] | ||
| m6A | Viral transcripts | n.a. | -Presence of viral m6A methylations at ISG20 binding position results in IFN-⍺-mediated viral mRNA degradation via ISG20-YTHDF2 complex | - | (+) Increased ISG activity | [114] | ||
| m6A | Viral transcripts | n.a. | -m6A methylation of HBx transcript mediated by its own protein leads to decreased mRNA stability via YTHDF2 binding | - | n.a. | [115] | ||
| Class I: dsDNA Viruses | ||||||||
| Viral Family | Virus Name | Chemical Modification/ Machinery | Analyzed Modified RNA | Detection Technique | Main Outcome(s) | Impact on Replication | Impact on Host Immune Response | Ref. |
| Adenoviridae | Fowl Adenovirus Serotype 4 (FAdV-4) | m6A | Host IFNB mRNA | m6A-seq | -Viral infection increases m6A methylation of IFN-I mRNA and leads to its destabilization | + | (-) Decreased IFN-I signaling | [109] |
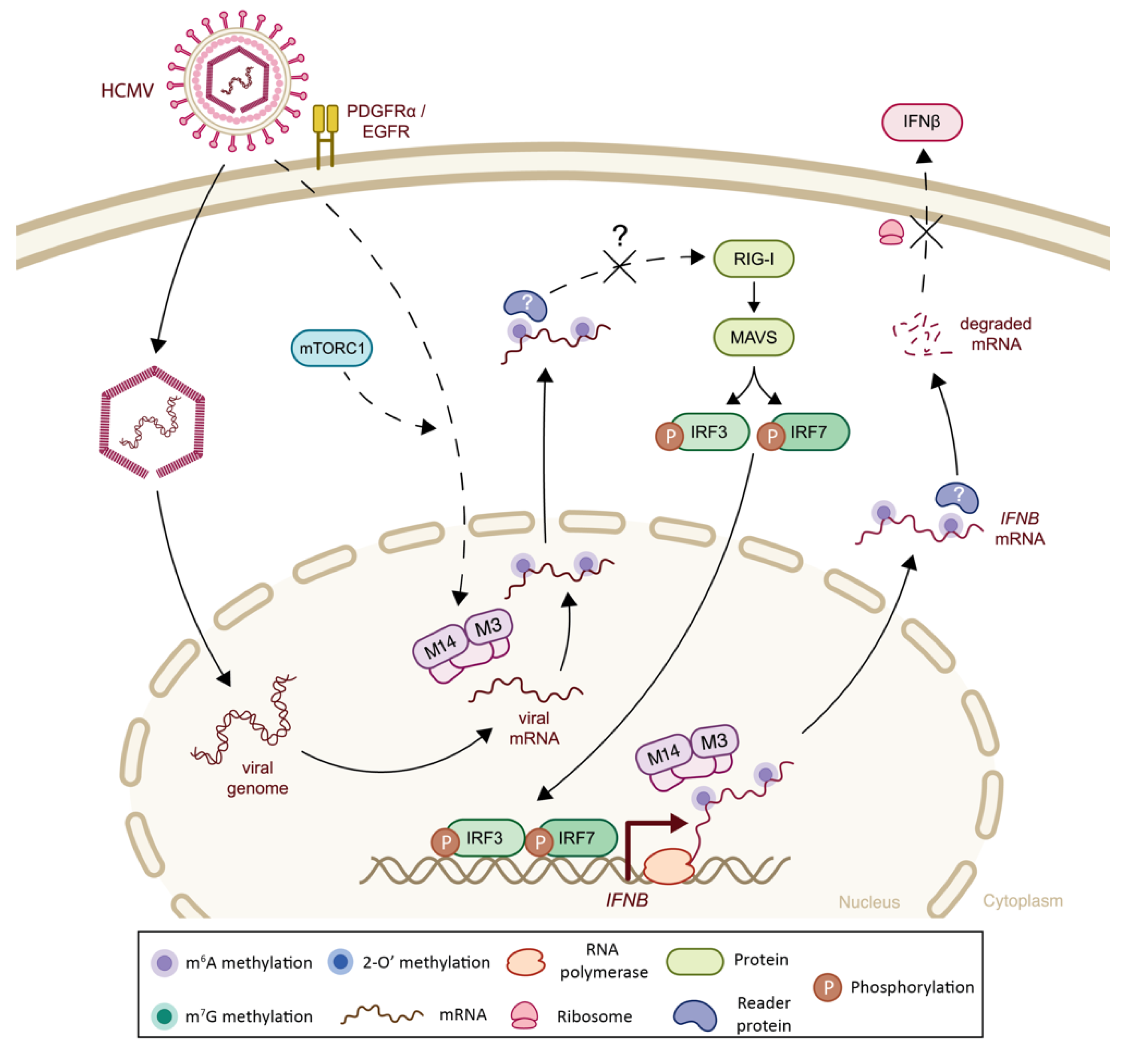
| Herpesviridae | Human Cytomegalovirus (HCMV) | m6A | Host IFNA and IFNB mRNA | m6A-seq | -Viral infection increases m6A methylation of IFNA and IFNB mRNAs, leading to transcript destabilization and subsequent impairment of signaling cascade Note: In vivo validation in murine model provided | + | (-) Decreased IFN-I signaling | [109] |
| m6A | Host IFNB mRNA | m6A-seq | -Viral infection increases the level of m6A machinery and induces IFNB mRNA methylation | + | (-) Decreased IFNβ signaling | [116] | ||
| Herpesviridae | Human Cytomegalovirus (HCMV) | m6A | n.a. | n.a. | -METTL3 acts as a negative regulator of the IFNβ innate immunity cascade in response to infection | n.a. | (-) Decreased IFNβ signaling | [106] |
| Herpes Simplex virus (HSV) | m6A | n.a. | n.a. | -METTL3 translocates to the cytoplasm and negatively regulates IFNβ innate immunity cascade in response to infection | n.a. | (-) Decreased IFNβ signaling | [106] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mersinoglu, B.; Cristinelli, S.; Ciuffi, A. The Impact of Epitranscriptomics on Antiviral Innate Immunity. Viruses 2022, 14, 1666. https://doi.org/10.3390/v14081666
Mersinoglu B, Cristinelli S, Ciuffi A. The Impact of Epitranscriptomics on Antiviral Innate Immunity. Viruses. 2022; 14(8):1666. https://doi.org/10.3390/v14081666
Chicago/Turabian StyleMersinoglu, Beril, Sara Cristinelli, and Angela Ciuffi. 2022. "The Impact of Epitranscriptomics on Antiviral Innate Immunity" Viruses 14, no. 8: 1666. https://doi.org/10.3390/v14081666
APA StyleMersinoglu, B., Cristinelli, S., & Ciuffi, A. (2022). The Impact of Epitranscriptomics on Antiviral Innate Immunity. Viruses, 14(8), 1666. https://doi.org/10.3390/v14081666