
Viral and Cellular Factors Contributing to the Hematogenous Dissemination of Human Cytomegalovirus via Polymorphonuclear Leukocytes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Viruses
2.3. Generation of HCMV Mutants
2.4. PMN-Mediated Transmission of Cell-Associated HCMV Mutants
2.5. Blocking of Cellular Adhesion Molecules during PMN-Mediated Transfer of HCMV Isolates
2.6. Effects of HCMV Entry Inhibitors on PMN-Mediated Spread
2.7. Indirect Immunofluorescence
2.8. Immunoblotting
2.9. Statistical Analysis
3. Results
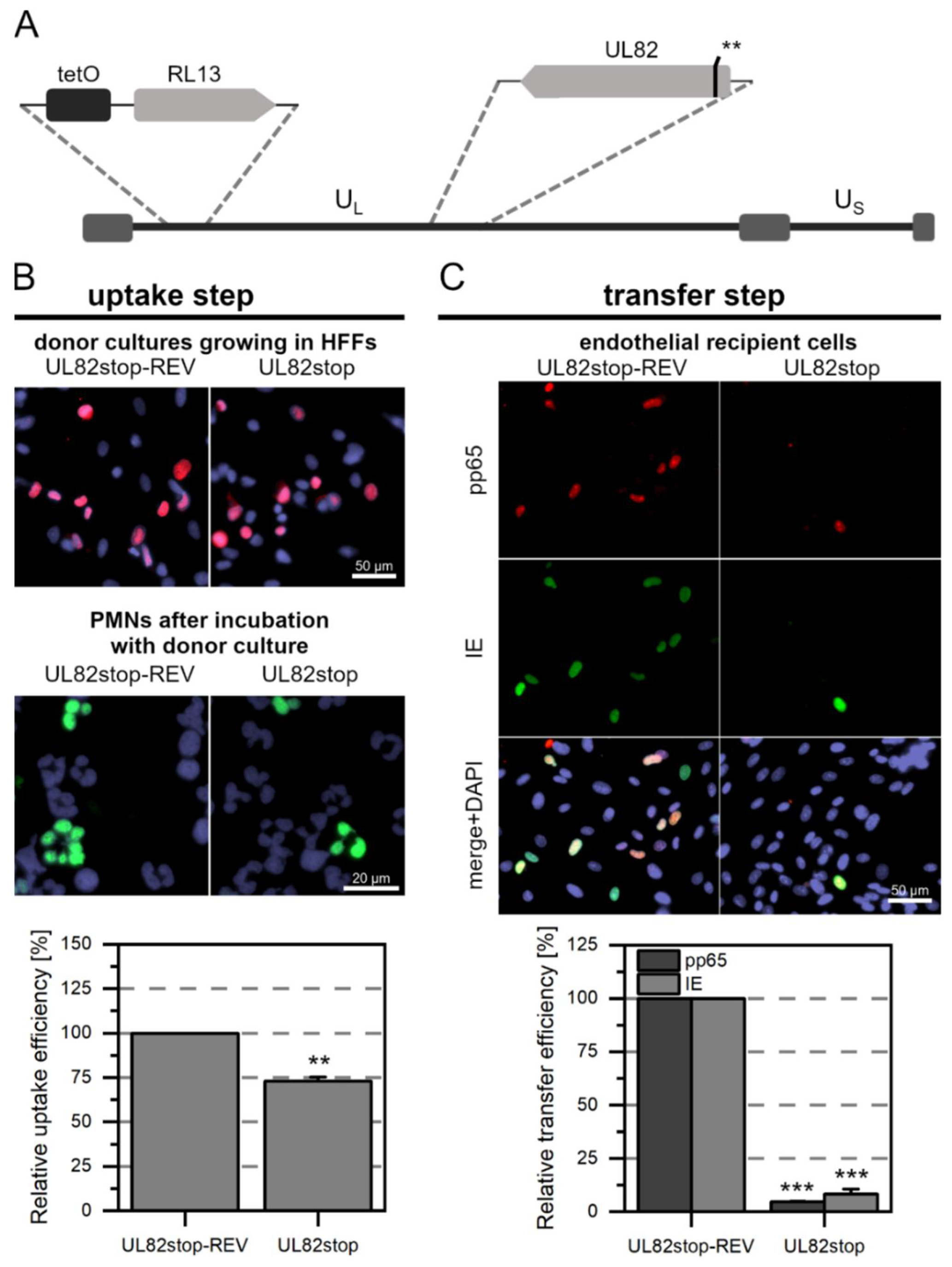
3.1. Generation of HCMV Mutants and Revertants Thereof
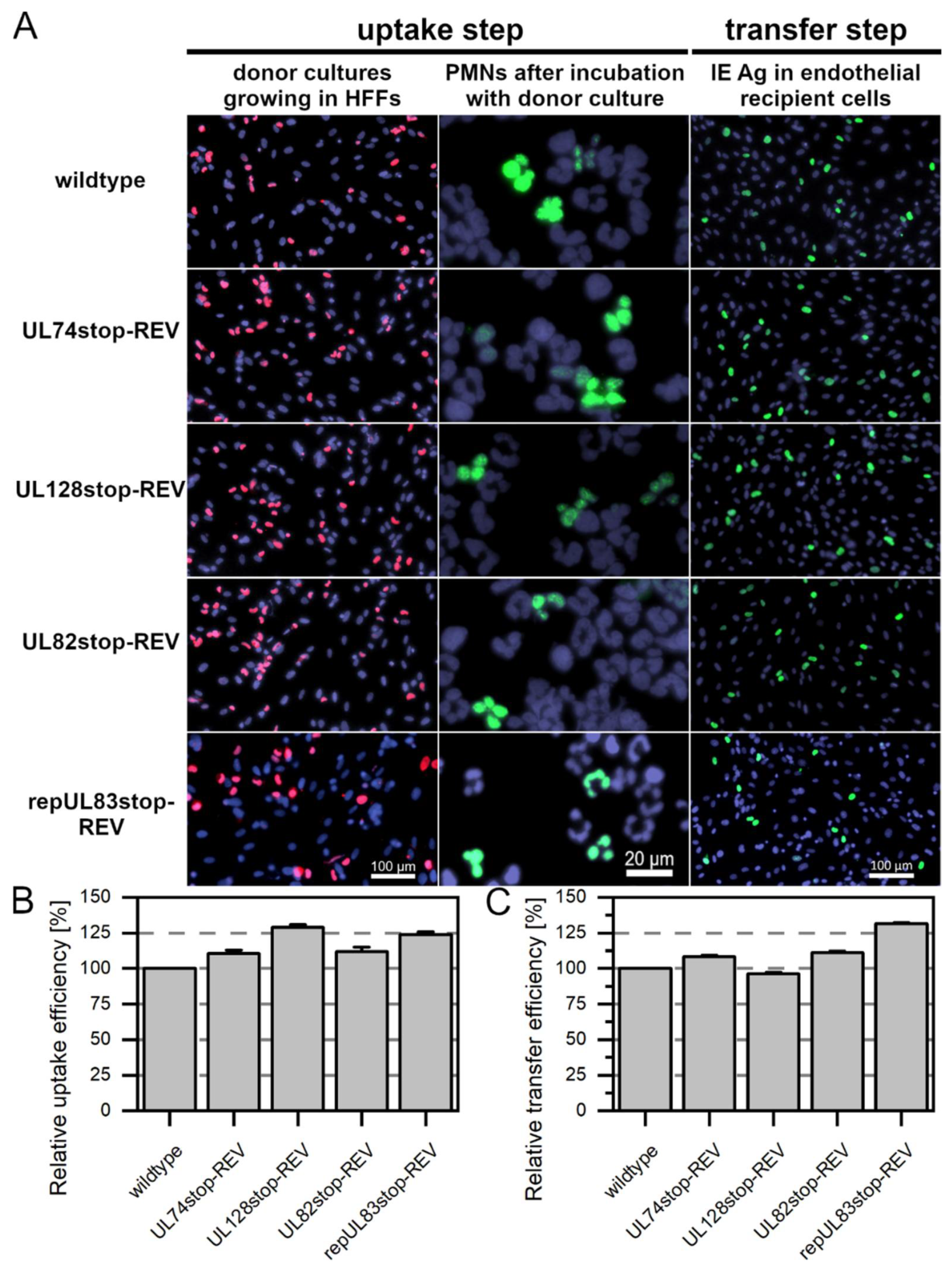
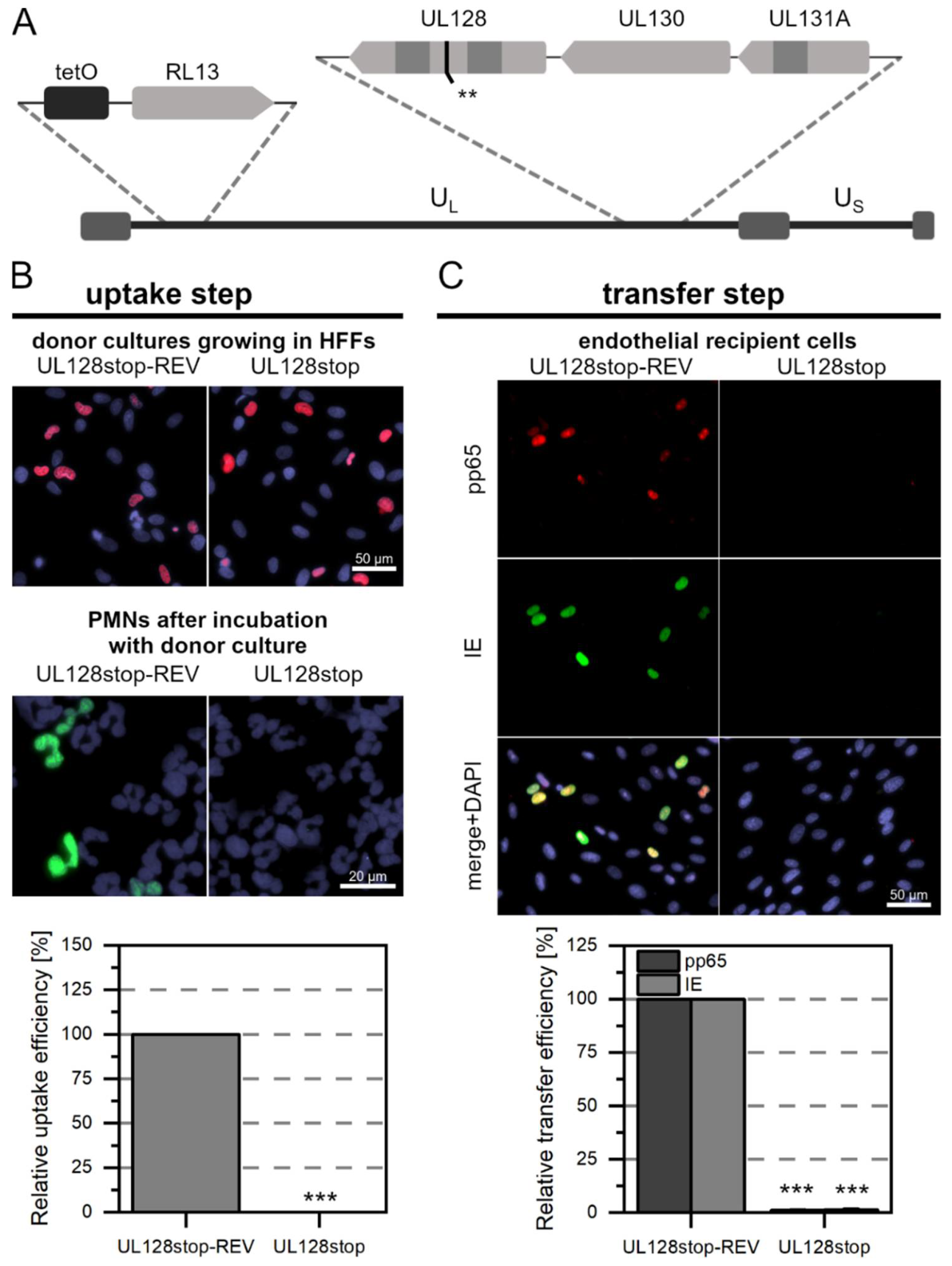
3.2. While pUL128 Is Essential for Uptake of HCMV by PMNs, Glycoprotein O Is Dispensable for Uptake and Transfer
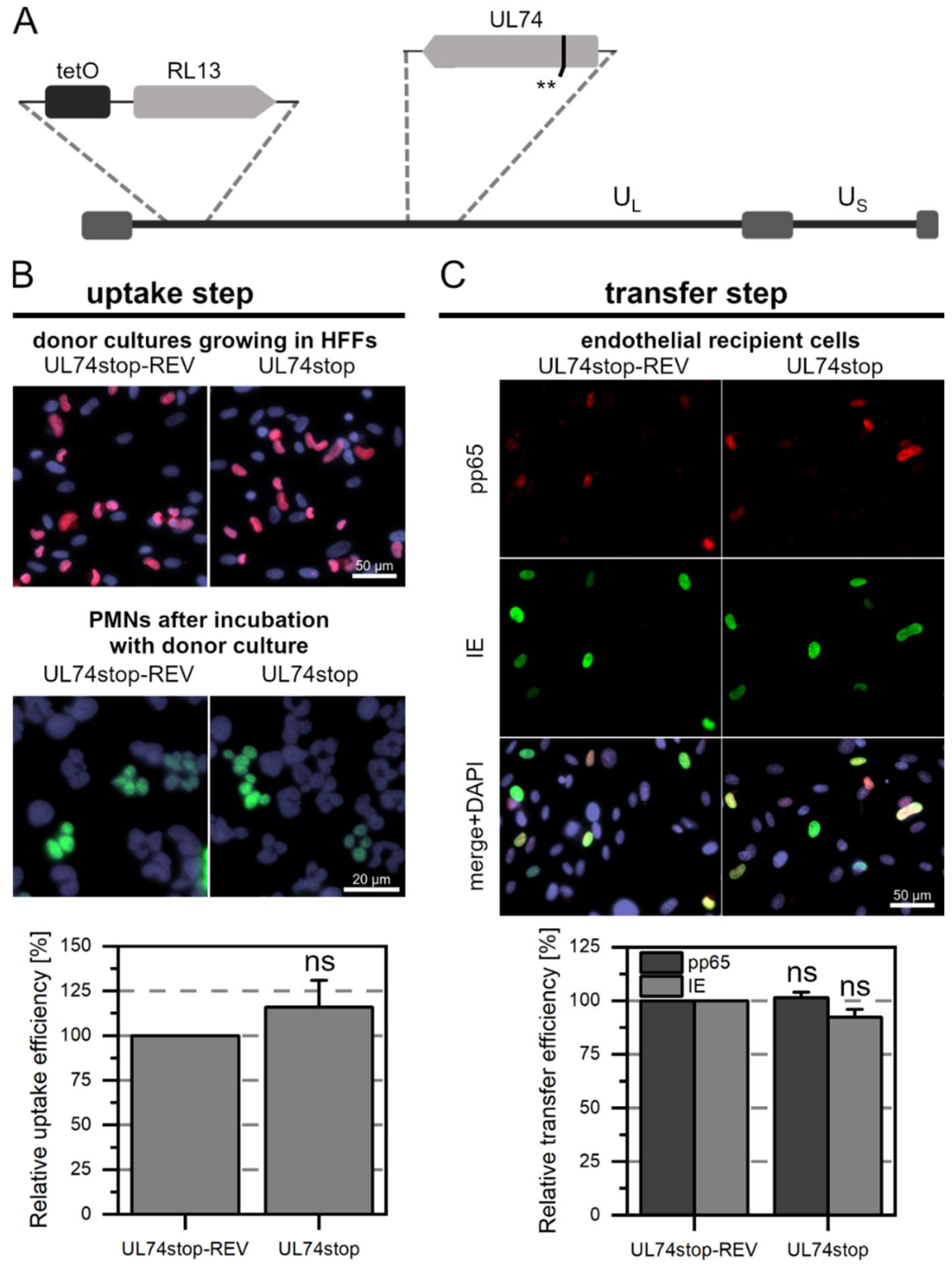
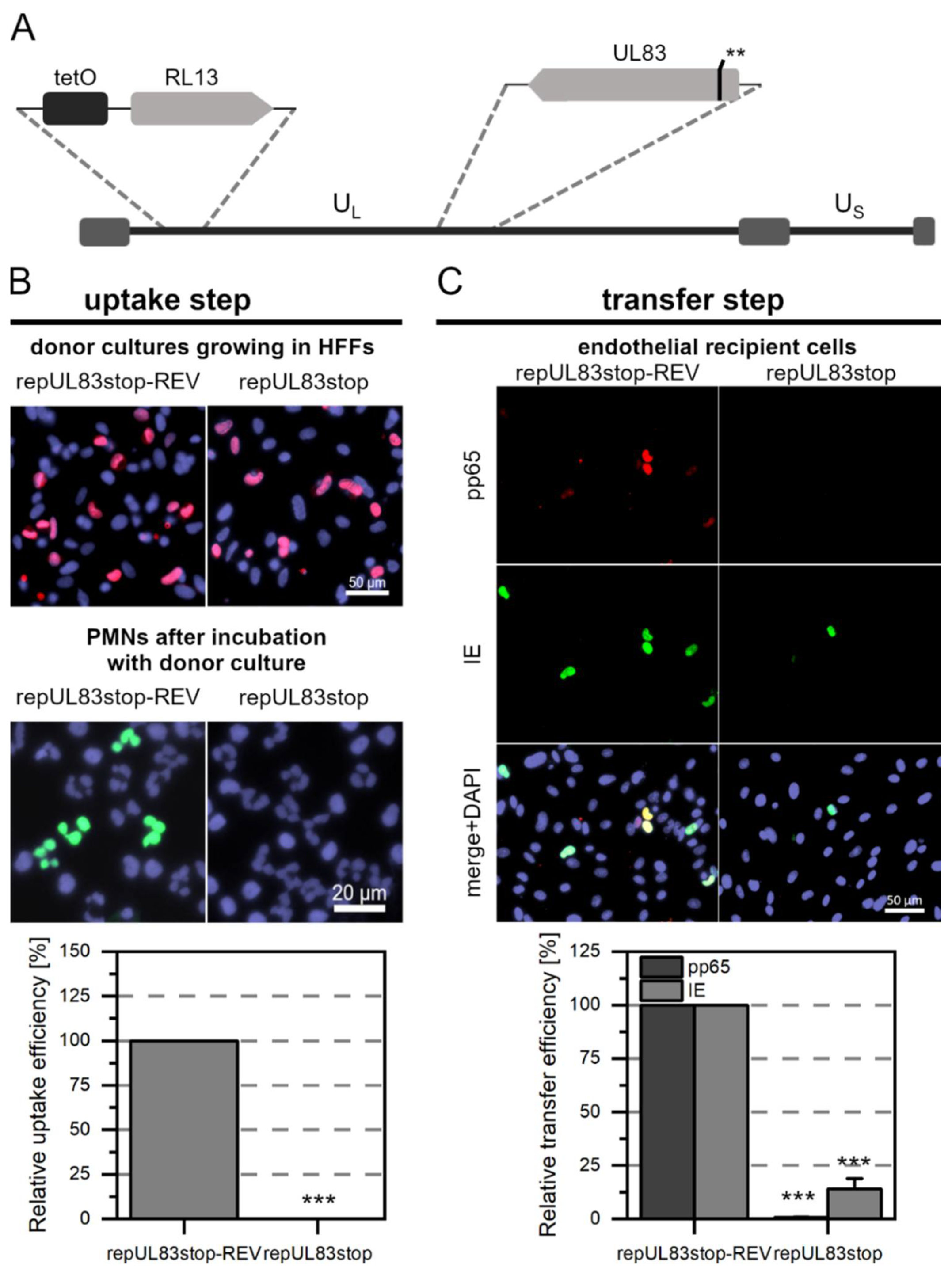
3.3. The Tegument Proteins pp65 and pp71 Contribute to PMN-Mediated Transmission of HCMV
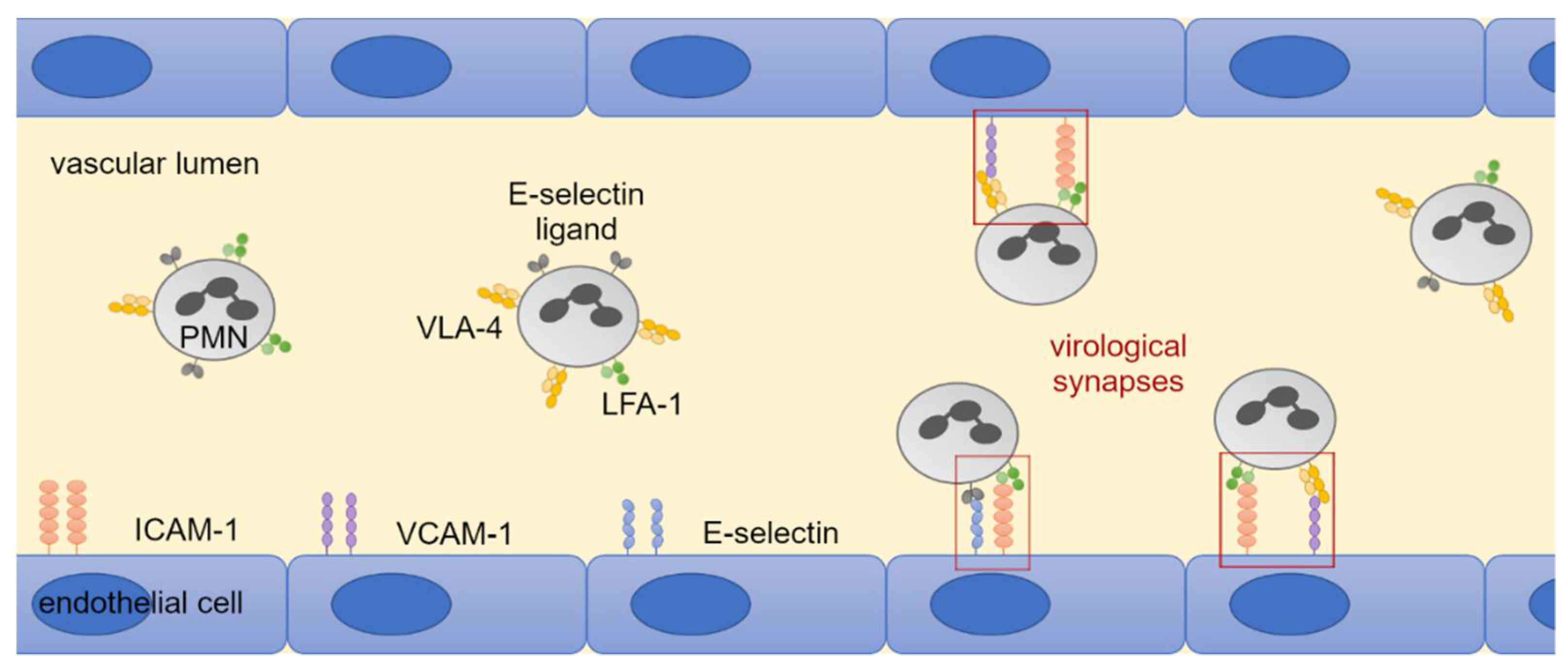
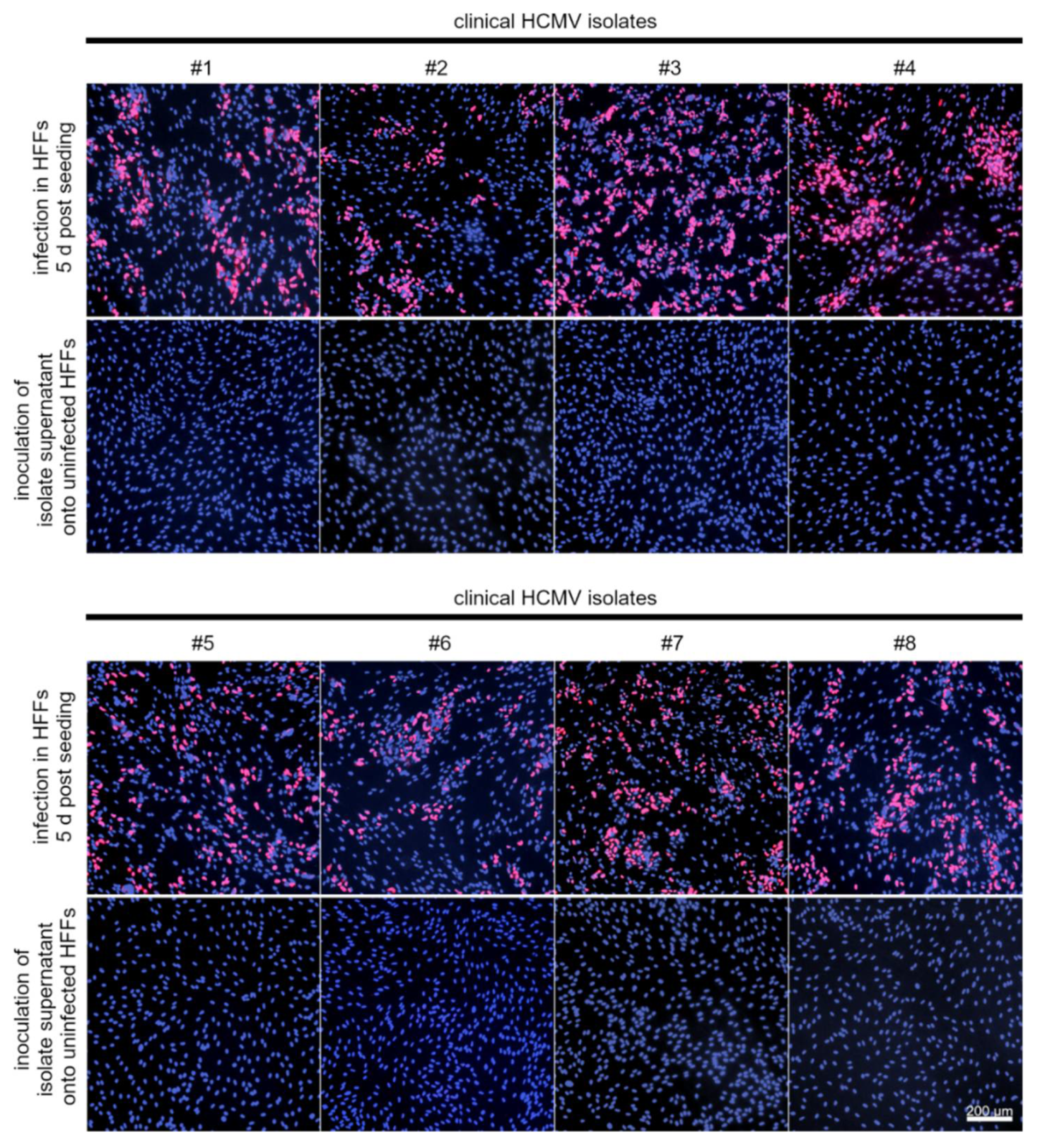
3.4. LFA-1 Is Required for Efficient Transfer of Cell-Associated HCMV Isolates via PMNs
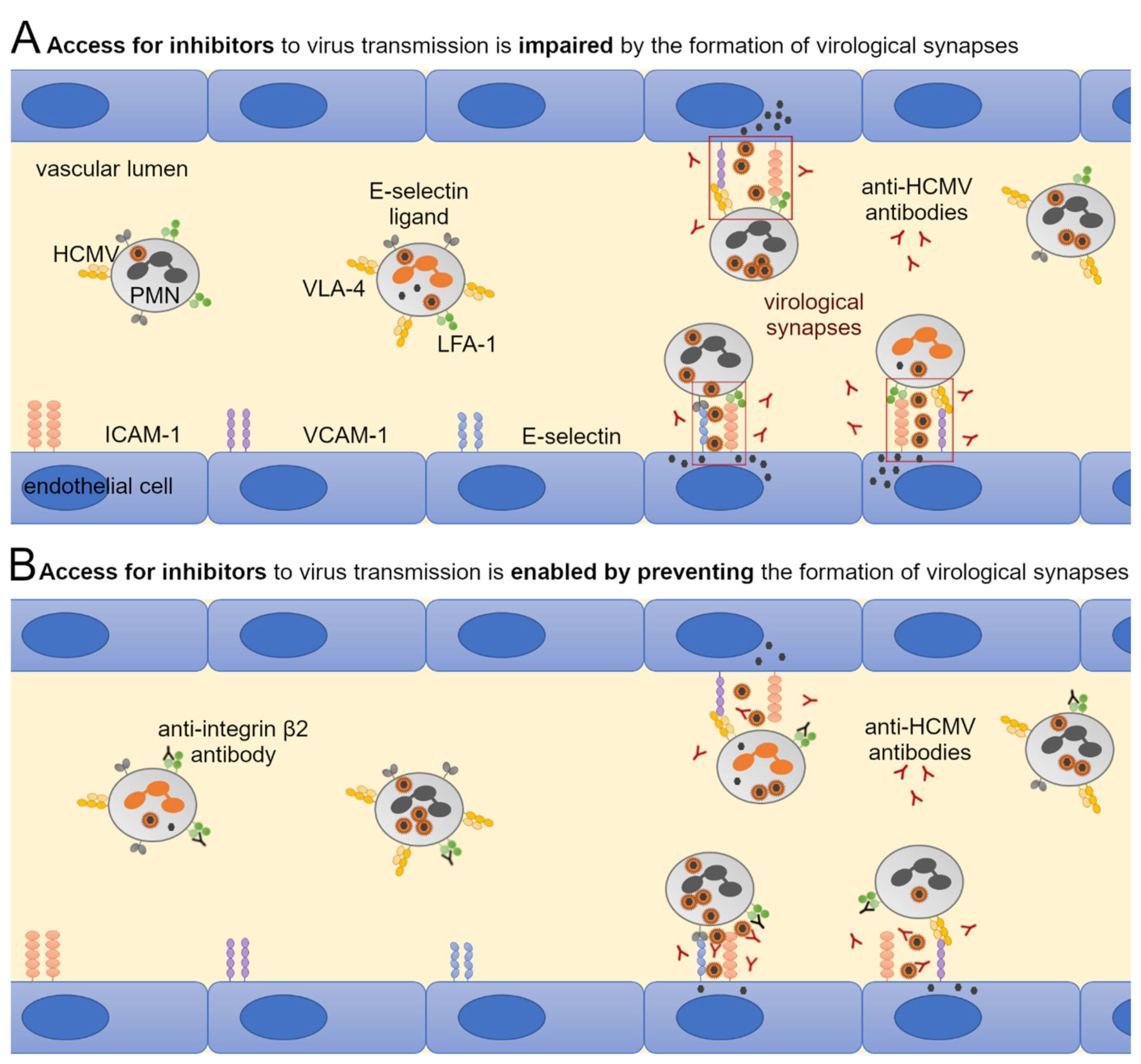
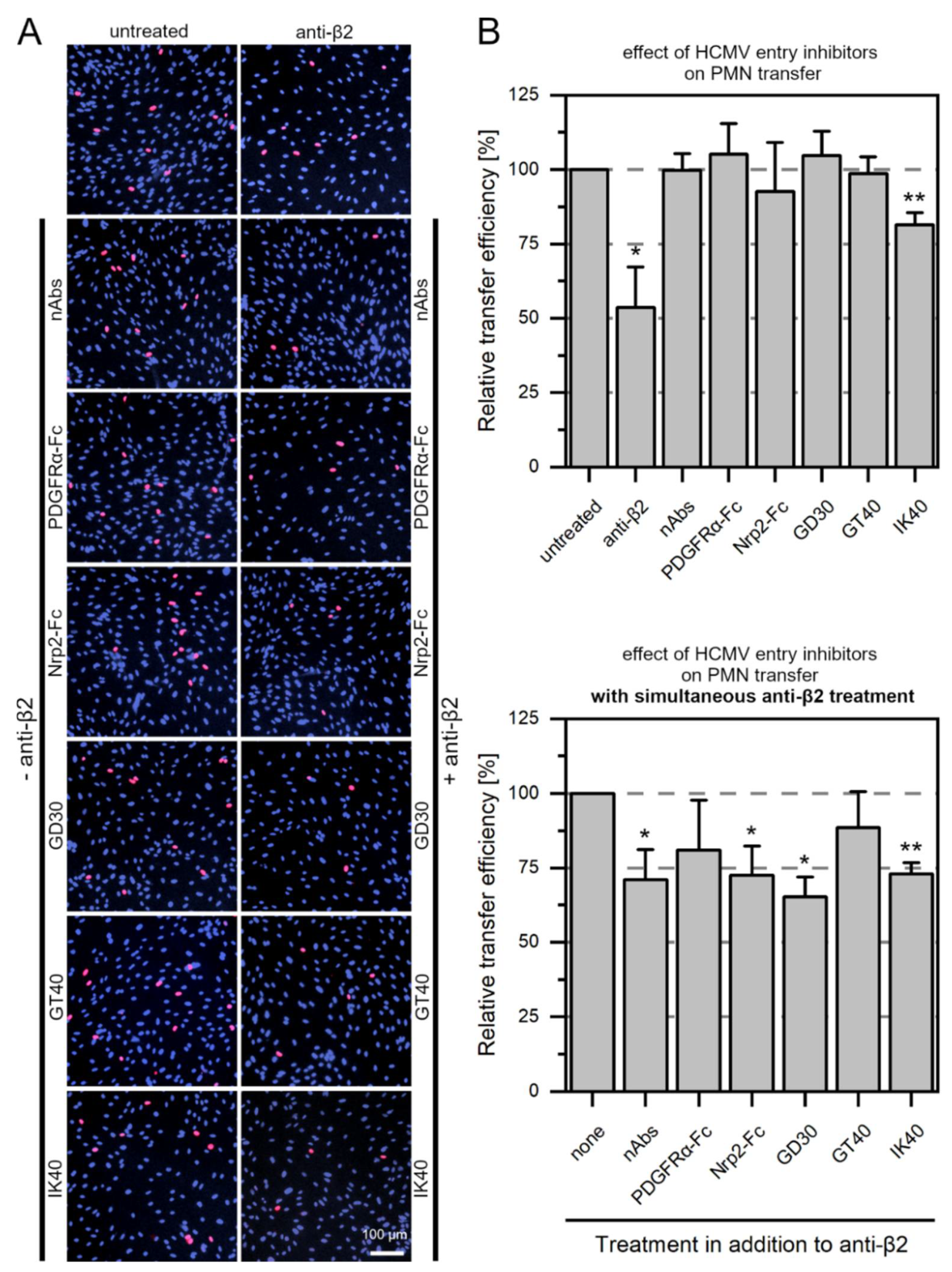
3.5. Large Entry Inhibitors Can Reduce PMN-Mediated Transfer of HCMV Clinical Isolates When the LFA-1 Subunit β2 Is Blocked
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.; Sinclair, J. Aspects of Human Cytomegalovirus Latency and Reactivation. Poxviruses 2008, 325, 297–313. [Google Scholar] [CrossRef]
- Gilbert, G.; Hudson, I.; Hayes, K.; James, J. Prevention of transfusion-acquired cytomegalovirus infection in infants by blood filtration to remove leucocytes. Neonatal Cytomegalovirus Infection Study Group. Lancet 1989, 333, 1228–1231. [Google Scholar] [CrossRef]
- Spector, S.A.; Merrill, R.; Wolf, D.; Dankner, W.M. Detection of human cytomegalovirus in plasma of AIDS patients during acute visceral disease by DNA amplification. J. Clin. Microbiol. 1992, 30, 2359–2365. [Google Scholar] [CrossRef] [Green Version]
- Grefte, A.; van der Giessen, M.; van Son, W.; The, T.H. Circulating cytomegalovirus (CMV)-infected endothelial cells in patients with an active CMV infection. J. Infect. Dis. 1993, 167, 270–277. [Google Scholar] [CrossRef]
- Sinzger, C.; Plachter, B.; Grefte, A.; The, T.H.; Jahn, G. Tissue Macrophages Are Infected by Human Cytomegalovirus In Vivo. J. Infect. Dis. 1996, 173, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Gerna, G.; Zipeto, D.; Percivalle, E.; Parea, M.; Revello, M.G.; Maccario, R.; Peri, G.; Milanesi, G. Human Cytomegalovirus Infection of the Major Leukocyte Subpopulations and Evidence for Initial Viral Replication in Polymorphonuclear Leukocytes from Viremic Patients. J. Infect. Dis. 1992, 166, 1236–1244. [Google Scholar] [CrossRef]
- Hassan-Walker, A.F.; Mattes, F.M.; Griffiths, P.D.; Emery, V.C. Quantity of cytomegalovirus DNA in different leukocyte populations during active infection in vivo and the presence of gB and UL18 transcripts. J. Med. Virol. 2001, 64, 283–289. [Google Scholar] [CrossRef]
- Grefte, A.; Harmsen, M.C.; Van Der Giessen, M.; Knollema, S.; Van Son, W.J.; The, T.H. Presence of human cytomegalovirus (HCMV) immediate early mRNA but not ppUL83 (lower matrix protein pp65) mRNA in polymorphonuclear and mononuclear leukocytes during active HCMV infection. J. Gen. Virol. 1994, 75 Pt 8, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Grefte, J.M.M.; Van Der Gun, B.T.F.; Schmolke, S.; Van Der Giessen, M.; Van Son, W.J.; Plachter, B.; Jahn, G.; The, T.H. Cytomegalovirus Antigenemia Assay: Identification of the Viral Antigen as the Lower Matrix Protein PP65. J. Infect. Dis. 1992, 166, 683–684. [Google Scholar] [CrossRef] [PubMed]
- Kas-Deelen, A.M.; The, T.H.; Blom, N.; van der Strate, B.W.; De Maar, E.F.; Smit, J.; van Son, W.J.; Harmsen, M.C. Uptake of pp65 in in vitro generated pp65-positive polymorphonuclear cells mediated by phagocytosis and cell fusion? Intervirology 2001, 44, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Percivalle, E.; Torsellini, M.; Revello, M.G. Standardization of the human cytomegalovirus antigenemia assay by means of in vitro-generated pp65-positive peripheral blood polymorphonuclear leukocytes. J. Clin. Microbiol. 1998, 36, 3585–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.M.; Rana, P.S.J.B.; Jaeger, H.K.; O’Dowd, J.M.; Balemba, O.B.; Fortunato, E.A. Human Cytomegalovirus Compromises Development of Cerebral Organoids. J. Virol. 2019, 93, e00957-19. [Google Scholar] [CrossRef] [Green Version]
- Rüger, B.; Klages, S.; Walla, B.; Albrecht, J.; Fleckenstein, B.; Tomlinson, P.; Barrell, B. Primary structure and transcription of the genes coding for the two virion phosphoproteins pp65 and pp71 of human cytomegalovirus. J. Virol. 1987, 61, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Bresnahan, W.A.; Shenk, T.E. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14506–14511. [Google Scholar] [CrossRef] [Green Version]
- Marshall, E.E.; Malouli, D.; Hansen, S.G.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; Selseth, A.N.; Ford, J.C.; Burke, D.; et al. Enhancing safety of cytomegalovirus-based vaccine vectors by engaging host intrinsic immunity. Sci. Transl. Med. 2019, 11, eaaw2603. [Google Scholar] [CrossRef]
- Gerna, G.; Percivalle, E.; Baldanti, F.; Sozzani, S.; Lanzarini, P.; Genini, E.; Lilleri, D.; Revello, M.G. Human Cytomegalovirus Replicates Abortively in Polymorphonuclear Leukocytes after Transfer from Infected Endothelial Cells via Transient Microfusion Events. J. Virol. 2000, 74, 5629–5638. [Google Scholar] [CrossRef] [Green Version]
- Braun, B.; Sinzger, C. Transmission of cell-associated human cytomegalovirus isolates between various cell types using polymorphonuclear leukocytes as a vehicle. Med. Microbiol. Immunol. 2021, 210, 197–209. [Google Scholar] [CrossRef]
- Yamane, Y.; Furukawa, T.; Plotkin, S.A. Supernatant virus release as a differentiating marker between low passage and vaccine strains of human cytomegalovirus. Vaccine 1983, 1, 23–25. [Google Scholar] [CrossRef]
- Sinzger, C.; Schmidt, K.; Knapp, J.; Kahl, M.; Beck, R.; Waldman, J.; Hebart, H.; Einsele, H.; Jahn, G. Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J. Gen. Virol. 1999, 80 Pt 11, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Dargan, D.J.; Douglas, E.; Cunningham, C.; Jamieson, F.; Stanton, R.; Baluchova, K.; McSharry, B.; Tomasec, P.; Emery, V.; Percivalle, E.; et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 2010, 91, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.; Neale, M.L.; Davies, J.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Investig. 2010, 120, 3191–3208. [Google Scholar] [CrossRef] [Green Version]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [Green Version]
- Adler, B.; Scrivano, L.; Ruzcics, Z.; Rupp, B.; Sinzger, C.; Koszinowski, U. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 2006, 87 Pt 9, 2451–2460. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. Characterization of the Human Cytomegalovirus gH/gL/UL128-131 Complex That Mediates Entry into Epithelial and Endothelial Cells. J. Virol. 2008, 82, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19. [Google Scholar] [CrossRef] [Green Version]
- Brait, N.; Stögerer, T.; Kalser, J.; Adler, B.; Kunz, I.; Benesch, M.; Kropff, B.; Mach, M.; Puchhammer-Stöckl, E.; Görzer, I. Influence of Human Cytomegalovirus Glycoprotein O Polymorphism on the Inhibitory Effect of Soluble Forms of Trimer- and Pentamer-Specific Entry Receptors. J. Virol. 2020, 94, e00107-20. [Google Scholar] [CrossRef]
- Kschonsak, M.; Johnson, M.C.; Schelling, R.; Green, E.M.; Rougé, L.; Ho, H.; Patel, N.; Kilic, C.; Kraft, E.; Arthur, C.P.; et al. Structural basis for HCMV Pentamer receptor recognition and antibody neutralization. Sci. Adv. 2022, 8, eabm2536. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, C.; Hochdorfer, D.; Lieber, D.; Subramanian, N.; Stöhr, D.; Sampaio, K.L.; Sinzger, C. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PLoS Pathog. 2017, 13, e1006273. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizić, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Braun, B.; Fischer, D.; Sampaio, K.L.; Mezger, M.; Stöhr, D.; Stanton, R.J.; Sinzger, C. Peptide Derivatives of Platelet-Derived Growth Factor Receptor Alpha Inhibit Cell-Associated Spread of Human Cytomegalovirus. Viruses 2021, 13, 1780. [Google Scholar] [CrossRef]
- Sinzger, C.; Mangin, M.; Weinstock, C.; Topp, M.S.; Hebart, H.; Einsele, H.; Jahn, G. Effect of serum and CTL on focal growth of human cytomegalovirus. J. Clin. Virol. 2007, 38, 112–119. [Google Scholar] [CrossRef]
- Falk, J.J.; Winkelmann, M.; Sampaio, K.L.; Paal, C.; Schrezenmeier, H.; Alt, M.; Stanton, R.; Krawczyk, A.; Lotfi, R.; Sinzger, C. Large-Scale Screening of HCMV-Seropositive Blood Donors Indicates that HCMV Effectively Escapes from Antibodies by Cell-Associated Spread. Viruses 2018, 10, 500. [Google Scholar] [CrossRef] [Green Version]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus Cell-to-Cell Transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef] [Green Version]
- Zhong, P.; Agosto, L.M.; Munro, J.B.; Mothes, W. Cell-to-cell transmission of viruses. Curr. Opin. Virol. 2013, 3, 44–50. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Wu, L.; Bohks, S.M.; KewalRamani, V.N.; Unutmaz, D.; Hope, T.J. Recruitment of HIV and Its Receptors to Dendritic Cell-T Cell Junctions. Science 2003, 300, 1295–1297. [Google Scholar] [CrossRef] [Green Version]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 Cell to Cell Transfer across an Env-induced, Actin-dependent Synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.C.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R.M. Spread of HTLV-I Between Lymphocytes by Virus-Induced Polarization of the Cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, M.; Yoon, M.; Sloan, D.D.; Spear, P.G.; Jerome, K.R. The Virological Synapse Facilitates Herpes Simplex Virus Entry into T Cells. J. Virol. 2009, 83, 6171–6183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, M.-D. Neutrophil transendothelial migration: Updates and new perspectives. Blood 2019, 133, 2149–2158. [Google Scholar] [CrossRef]
- Revello, M.G.; Percivalle, E.; Arbustini, E.; Pardi, R.; Sozzani, S.; Gerna, G. In vitro generation of human cytomegalovirus pp65 antigenemia, viremia, and leukoDNAemia. J. Clin. Investig. 1998, 101, 2686–2692. [Google Scholar] [CrossRef] [Green Version]
- May, T.; Butueva, M.; Bantner, S.; Markusic, D.; Seppen, J.; MacLeod, R.A.; Weich, H.; Hauser, H.; Wirth, D. Synthetic Gene Regulation Circuits for Control of Cell Expansion. Tissue Eng. Part. A 2010, 16, 441–452. [Google Scholar] [CrossRef]
- Lieber, D.; Hochdorfer, D.; Stoehr, D.; Schubert, A.; Lotfi, R.; May, T.; Wirth, D.; Sinzger, C. A permanently growing human endothelial cell line supports productive infection with human cytomegalovirus under conditional cell growth arrest. BioTechniques 2015, 59, 127–136. [Google Scholar] [CrossRef]
- Laib Sampaio, K.; Stegmann, C.; Brizic, I.; Adler, B.; Stanton, R.J.; Sinzger, C. The contribution of pUL74 to growth of human cytomegalovirus is masked in the presence of RL13 and UL128 expression. J. Gen. Virol. 2016, 97, 1917–1927. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En Passant Mutagenesis: A Two Step Markerless Red Recombination System. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [CrossRef]
- Murrell, I.; Wilkie, G.S.; Davison, A.J.; Statkute, E.; Fielding, C.A.; Tomasec, P.; Wilkinson, G.W.G.; Stanton, R.J. Genetic Stability of Bacterial Artificial Chromosome-Derived Human Cytomegalovirus during Culture In Vitro. J. Virol. 2016, 90, 3929–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laib Sampaio, K.; Lutz, C.; Engels, R.; Stöhr, D.; Sinzger, C. Selection of Human Cytomegalovirus Mutants with Resistance against PDGFRα-Derived Entry Inhibitors. Viruses. 2021, 13, 1094. [Google Scholar] [CrossRef] [PubMed]
- Waldo, F.B.; Tomana, M.; Britt, W.; Julian, B.; Mestecky, J. Non-specific mesangial staining with antibodies against cytomegalovirus in immunoglobulin—A nephropathy. Lancet 1989, 333, 129–131. [Google Scholar] [CrossRef]
- Gebert, S.; Schmolke, S.; Sorg, G.; Flöss, S.; Plachter, B.; Stamminger, T. The UL84 protein of human cytomegalovirus acts as a transdominant inhibitor of immediate-early-mediated transactivation that is able to prevent viral replication. J. Virol. 1997, 71, 7048–7060. [Google Scholar] [CrossRef] [Green Version]
- Kalejta, R.F.; Bechtel, J.T.; Shenk, T. Human Cytomegalovirus pp71 Stimulates Cell Cycle Progression by Inducing the Proteasome-Dependent Degradation of the Retinoblastoma Family of Tumor Suppressors. Mol. Cell. Biol. 2003, 23, 1885–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreoni, M.; Faircloth, M.; Vugler, L.; Britt, W.J. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 1989, 23, 157–167. [Google Scholar] [CrossRef]
- Sanchez, V.; Greis, K.D.; Sztul, E.; Britt, W.J. Accumulation of Virion Tegument and Envelope Proteins in a Stable Cytoplasmic Compartment during Human Cytomegalovirus Replication: Characterization of a Potential Site of Virus Assembly. J. Virol. 2000, 74, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Kalejta, R.F.; Albright, E.R. Expanding the Known Functional Repertoire of the Human Cytomegalovirus pp71 Protein. Front. Cell. Infect. Microbiol. 2020, 10, 95. [Google Scholar] [CrossRef] [Green Version]
- Sinzger, C.; Müntefering, H.; Löning, T.; Stöss, H.; Plachter, B.; Jahn, G. Cell types infected in human cytomegalovirus placentitis identified by immunohistochemical double staining. Virchows. Arch. A Pathol. Anat. Histopathol. 1993, 423, 249–256. [Google Scholar] [CrossRef]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.H.; The, T.H.; Jahn, G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 1995, 76 Pt 4, 741–750. [Google Scholar] [CrossRef]
- Bissinger, A.; Sinzger, C.; Kaiserling, E.; Jahn, G. Human cytomegalovirus as a direct pathogen: Correlation of multiorgan involvement and cell distribution with clinical and pathological findings in a case of congenital inclusion disease. J. Med. Virol. 2002, 67, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.; Sinzger, C. Endothelial cells in human cytomegalovirus infection: One host cell out of many or a crucial target for virus spread? Thromb. Haemost. 2009, 102, 1057–1063. [Google Scholar] [PubMed] [Green Version]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus Cell Tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [CrossRef] [PubMed]
- Lemmermann, N.; Krmpotic, A.; Podlech, J.; Brizić, I.; Prager, A.; Adler, H.; Karbach, A.; Wu, Y.; Jonjic, S.; Reddehase, M.; et al. Non-redundant and Redundant Roles of Cytomegalovirus gH/gL Complexes in Host Organ Entry and Intra-tissue Spread. PLoS Pathog. 2015, 11, e1004640. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.T.; Compton, T. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 1998, 72, 8191–8197. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Nelson, J.A.; Britt, W.J. Glycoprotein H-related complexes of human cytomegalovirus: Identification of a third protein in the gCIII complex. J. Virol. 1997, 71, 3090–3097. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lanchy, J.-M.; Ryckman, B.J. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Shinoda, T.; Shirasago, Y.; Kondoh, M.; Shinya, N.; Hanada, K.; Yagi, K.; Suzuki, T.; Wakita, T.; Kimura-Someya, T.; et al. Occludin-binding single-chain variable fragment and antigen-binding fragment antibodies prevent hepatitis C virus infection. FEBS Lett. 2020, 595, 220–229. [Google Scholar] [CrossRef]
- Mathieu, C.; Ferren, M.; Harder, O.; Bovier, F.T.; Marcink, T.C.; Predella, C.; Angius, F.; Drew-Bear, J.; Dorrello, N.V.; Greninger, A.L.; et al. Single-chain variable fragment antibody constructs neutralize measles virus infection in vitro and in vivo. Cell. Mol. Immunol. 2021, 18, 1835–1837. [Google Scholar] [CrossRef]
- van Dorsten, R.T.; Reh, L.; Trkola, A.; Morris, L.; Moore, P.L. Single-Chain Variable Fragments of Broadly Neutralizing Antibodies Prevent HIV Cell-Cell Transmission. J. Virol. 2022, 96, e0193421. [Google Scholar] [CrossRef]
- Moazen, B.; Ebrahimi, E.; Nejatollahi, F. Single Chain Antibodies Against gp55 of Human Cytomegalovirus (HCMV) for Prophylaxis and Treatment of HCMV Infections. Jundishapur. J. Microbiol. 2016, 9, e16241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Ye, X.; Freed, D.C.; Li, L.; Ku, Z.; Xiong, W.; Gao, P.; Liu, X.; Montgomery, D.; Xu, W.; et al. Potent Bispecific Neutralizing Antibody Targeting Glycoprotein B and the gH/gL/pUL128/130/131 Complex of Human Cytomegalovirus. Antimicrob. Agents Chemother. 2021, 65, e02422-20. [Google Scholar] [CrossRef] [PubMed]
- Weiler, N.; Paal, C.; Adams, K.; Calcaterra, C.; Fischer, D.; Stanton, R.; Stöhr, D.; Sampaio, K.L.; Sinzger, C. Role of Envelope Glycoprotein Complexes in Cell-Associated Spread of Human Cytomegalovirus. Viruses 2021, 13, 614. [Google Scholar] [CrossRef] [PubMed]
- Podlech, J.; Reddehase, M.; Adler, B.; Lemmermann, N.A.W. Principles for studying in vivo attenuation of virus mutants: Defining the role of the cytomegalovirus gH/gL/gO complex as a paradigm. Med. Microbiol. Immunol. 2015, 204, 295–305. [Google Scholar] [CrossRef]
- Yunis, J.; Farrell, H.E.; Bruce, K.; Lawler, C.; Wyer, O.; Davis-Poynter, N.; Brizić, I.; Jonjić, S.; Adler, B.; Stevenson, P.G. Murine Cytomegalovirus Glycoprotein O Promotes Epithelial Cell Infection In Vivo. J. Virol. 2019, 93, e01378-18. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional Interaction between the pp71 Protein of Human Cytomegalovirus and the PML-Interacting Protein Human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef] [Green Version]
- Baldick, C.J.; Marchini, A.; Patterson, C.E.; Shenk, T. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 1997, 71, 4400–4408. [Google Scholar] [CrossRef] [Green Version]
- Reyda, S.; Tenzer, S.; Navarro, P.; Gebauer, W.; Saur, M.; Krauter, S.; Büscher, N.; Plachter, B. The Tegument Protein pp65 of Human Cytomegalovirus Acts as an Optional Scaffold Protein That Optimizes Protein Uploading into Viral Particles. J. Virol. 2014, 88, 9633–9646. [Google Scholar] [CrossRef] [Green Version]
- Schleiss, M.R.; Buus, R.J.; Choi, K.Y.; McGregor, A. An attenuated CMV vaccine with a deletion in tegument protein GP83 (pp65 homolog) protects against placental infection and improves pregnancy outcome in a Guinea pig challenge model. Future Virol. 2013, 8, 1151–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) |
|---|---|
| Merlin-UL82stop for | GTCTCAGGCATCGTCCTCGCCCGGTGAGGGACCCTCGTCGTAAGCAGCAGCAATTAGTTAAGCCGAAGCCGCCAGCGGAAGaggatgacgacgataagt |
| Merlin-UL82stop rev | GGCAGTGCAGGCGACCAAAGCTTCCGCTGGCGGCTTCGGCTTAACTAATTGCTGCTGCTTACGACGAGGGTCCCTCACCGGcaaccaattaaccaattctga |
| Merlin-UL82stop-REV for | GTCTCAGGCATCGTCCTCGCCCGGTGAGGGACCCTCGTCAGAAGCAGCAGCAATTAGTGAAGCAGAAGCCGCCAGCGGAAGaggatgacgacgataagt |
| Merlin-UL82stop-REV rev | GGCAGTGCAGGCGACCAAAGCTTCCGCTGGCGGCTTCTGCTTCACTAATTGCTGCTGCTTCTGACGAGGGTCCCTCACCGGcaaccaattaaccaattctga |
| Merlin-UL82stop short for | GTCTCAGGCATCGTCCTCGC |
| Merlin-UL83stop for | CGCAGGCAGCATGGAGTCGCGCGGTCGCCGTTGTCCAGAATAGATATCCGTACTGTGACCCATTTCGGGGCACGTGCTaggatgacgacgataagtaggg |
| Merlin-UL83stop rev | CGCGACTAAACACTGCTTTCAGCACGTGCCCCGAAATGGGTCACAGTACGGATATCTATTCTGGACAACGGCGACCGCcaaccaattaaccaattctgattag |
| Merlin-UL83stop-REV for | CGCAGGCAGCATGGAGTCGCGCGGTCGCCGTTGTCCAGAAATGATATCCGTACTTGGTCCTATTTCGGGGCACGTGCTaggatgacgacgataagtaggg |
| Merlin-UL83stop-REV rev | CGCGACTAAACACGGCTTTCAGCACGTGCCCCGAAATAGGACCAAGTACGGATATCATTTCTGGACAACGGCGACCGCcaaccaattaaccaattctgattag |
| Merlin-UL83stop short for | CGCAGGCAGCATGGAGTCGC |
| repMerlin-UL83stop for | CGCAGGCAGCATGGAGTCGCGCGGTCGCCGTTGTCCCGAATAGATATCCGTACTGTGACCCATTTCGGGGCACGTGCTaggatgacgacgataagt |
| repMerlin-UL83stop1 rev | TCACAGTACGGATATCTATTCGGGACAACGGCGACCGCcaaccaattaaccaattctga |
| repMerlin-UL83stop2 rev | CGCGACTAAACACGGCTTTCAGCACGTGCCCCGAAATGGGTCACAGTACGGATATCTA |
| repMerlin-UL83stopREV1 for | ATGATATCCGTACTGGGTCCCATTTCGGGGCACGTGCTaggatgacgacgataagt |
| repMerlin-UL83stopREV2 for | CGCAGGCAGCATGGAGTCGCGCGGTCGCCGTTGTCCCGAAATGATATCCGTACTGGGT |
| repMerlin-UL83stopREV1 rev | ACCCAGTACGGATATCATTTCGGGACAACGGCGACCGCcaaccaattaaccaattctga |
| repMerlin-UL83stopREV2 rev | CGCGACTAAACACGGCTTTCAGCACGTGCCCCGAAATGGGACCCAGTACGGATATCAT |
| Kanamycin universal rev | CAACCAATTAACCAATTCTGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, B.; Laib Sampaio, K.; Kuderna, A.K.; Widmann, M.; Sinzger, C. Viral and Cellular Factors Contributing to the Hematogenous Dissemination of Human Cytomegalovirus via Polymorphonuclear Leukocytes. Viruses 2022, 14, 1561. https://doi.org/10.3390/v14071561
Braun B, Laib Sampaio K, Kuderna AK, Widmann M, Sinzger C. Viral and Cellular Factors Contributing to the Hematogenous Dissemination of Human Cytomegalovirus via Polymorphonuclear Leukocytes. Viruses. 2022; 14(7):1561. https://doi.org/10.3390/v14071561
Chicago/Turabian StyleBraun, Berenike, Kerstin Laib Sampaio, Anna K. Kuderna, Miriam Widmann, and Christian Sinzger. 2022. "Viral and Cellular Factors Contributing to the Hematogenous Dissemination of Human Cytomegalovirus via Polymorphonuclear Leukocytes" Viruses 14, no. 7: 1561. https://doi.org/10.3390/v14071561
APA StyleBraun, B., Laib Sampaio, K., Kuderna, A. K., Widmann, M., & Sinzger, C. (2022). Viral and Cellular Factors Contributing to the Hematogenous Dissemination of Human Cytomegalovirus via Polymorphonuclear Leukocytes. Viruses, 14(7), 1561. https://doi.org/10.3390/v14071561