
A Novel Flavi-like Virus in Alfalfa (Medicago sativa L.) Crops along the Snake River Valley


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Alfalfa Sampling and Sample Processing
2.2. Mechanical Inoculations of Nicotiana benthamiana and Common Bean Plants
2.3. RNA Extraction and HTS Analysis
2.4. Nucleic Acid Extraction, RT-PCR Testing, and Sanger Sequencing
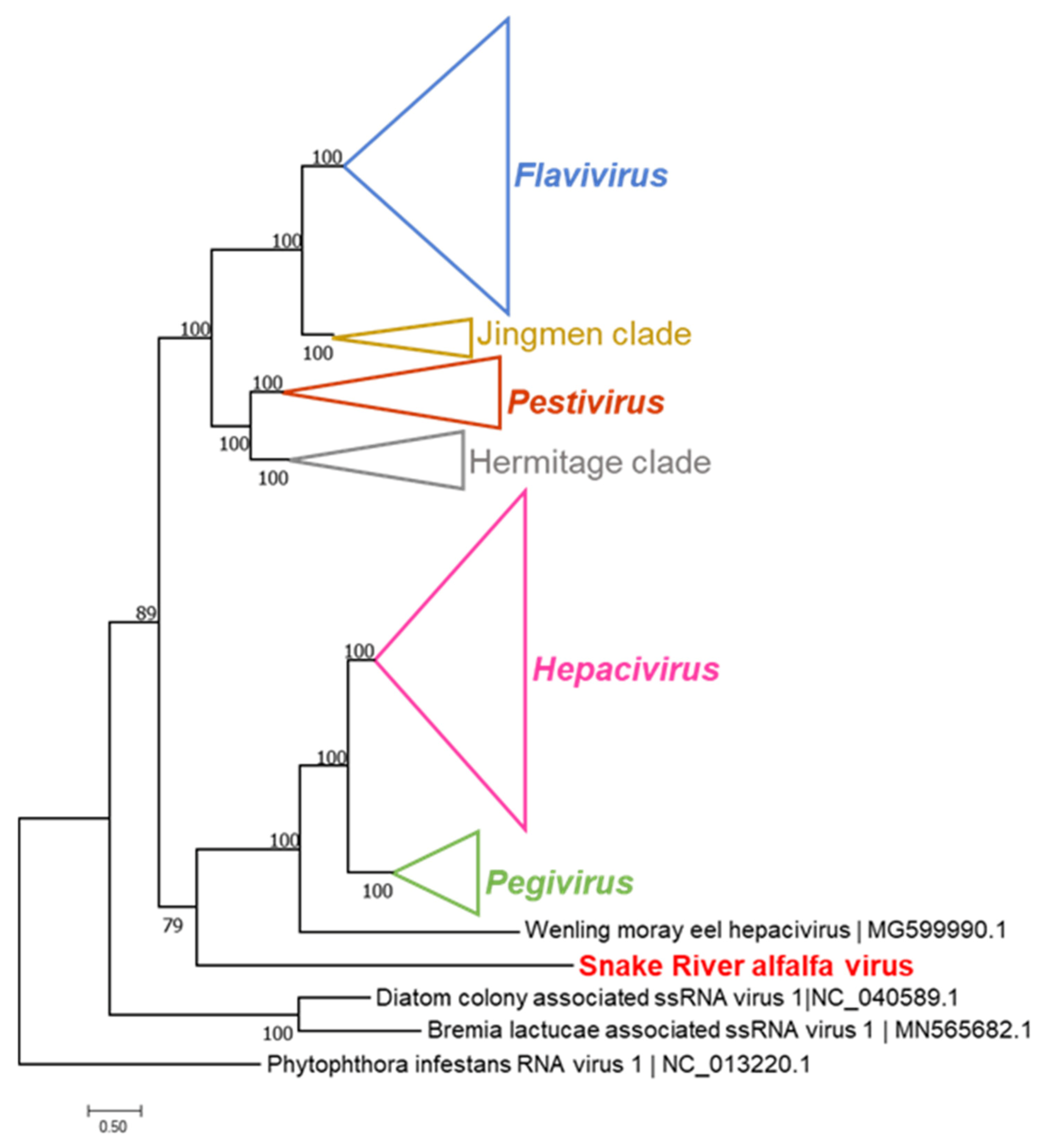
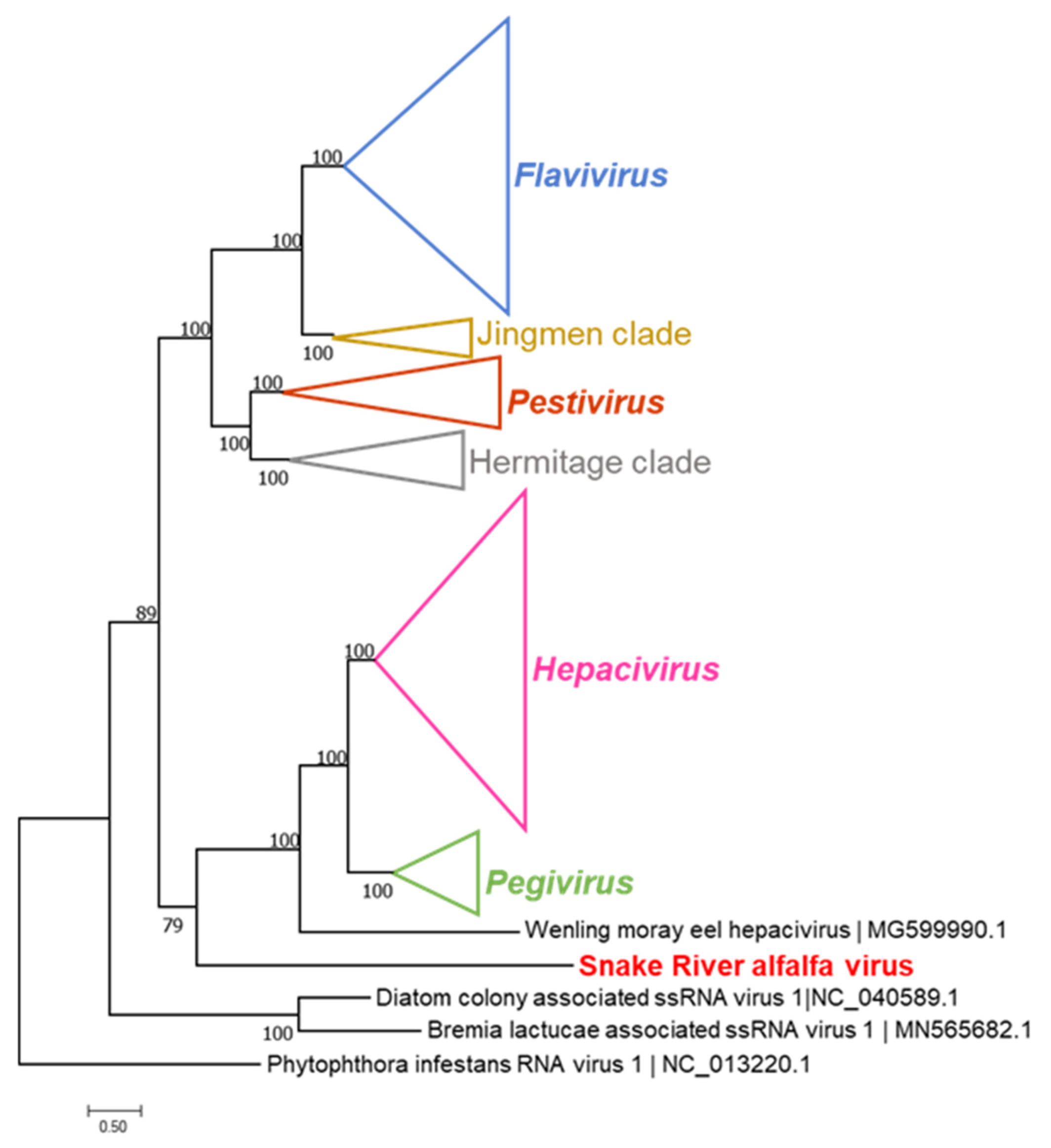
2.5. Sequence and Phylogenetic Analysis
3. Results
3.1. Viruses Identified in the 2020 Alfalfa Crop by High-Throughput Sequencing
3.2. SRAV Is Abundant in Alfalfa Stands of Different Ages in Southern Idaho
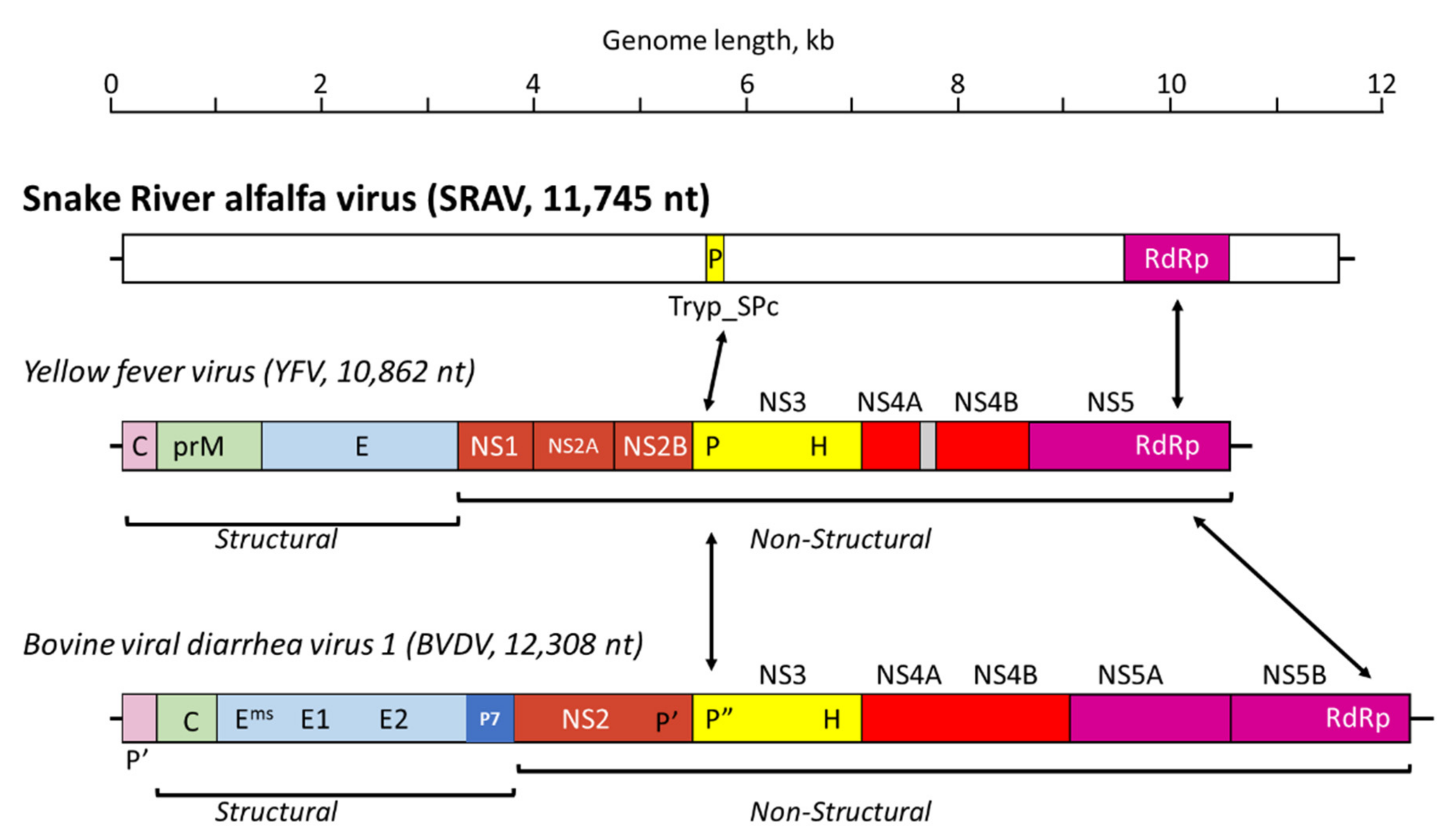
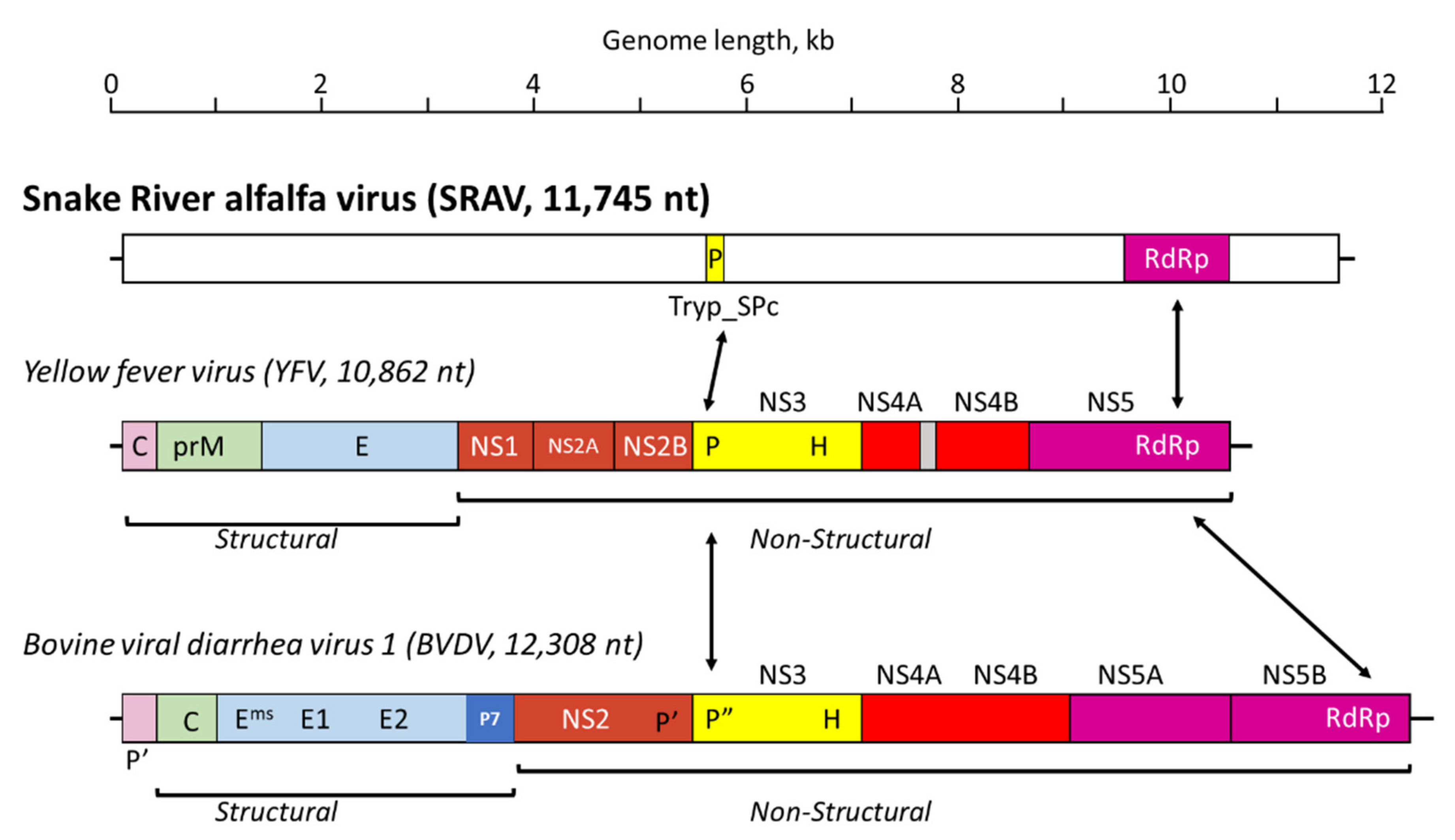
3.3. Sanger Resequencing of the SRAV Genomes, Acquisition of the 5′- and 3′-Termini, and Sequence Analysis
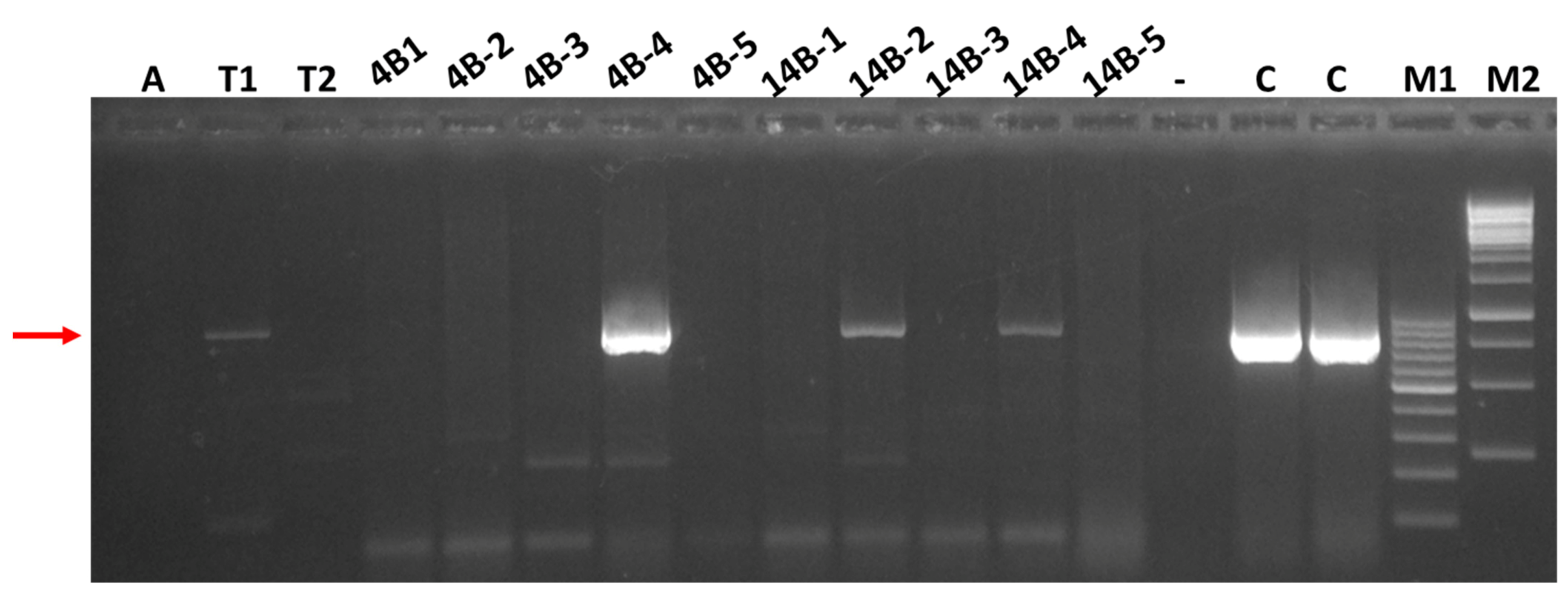
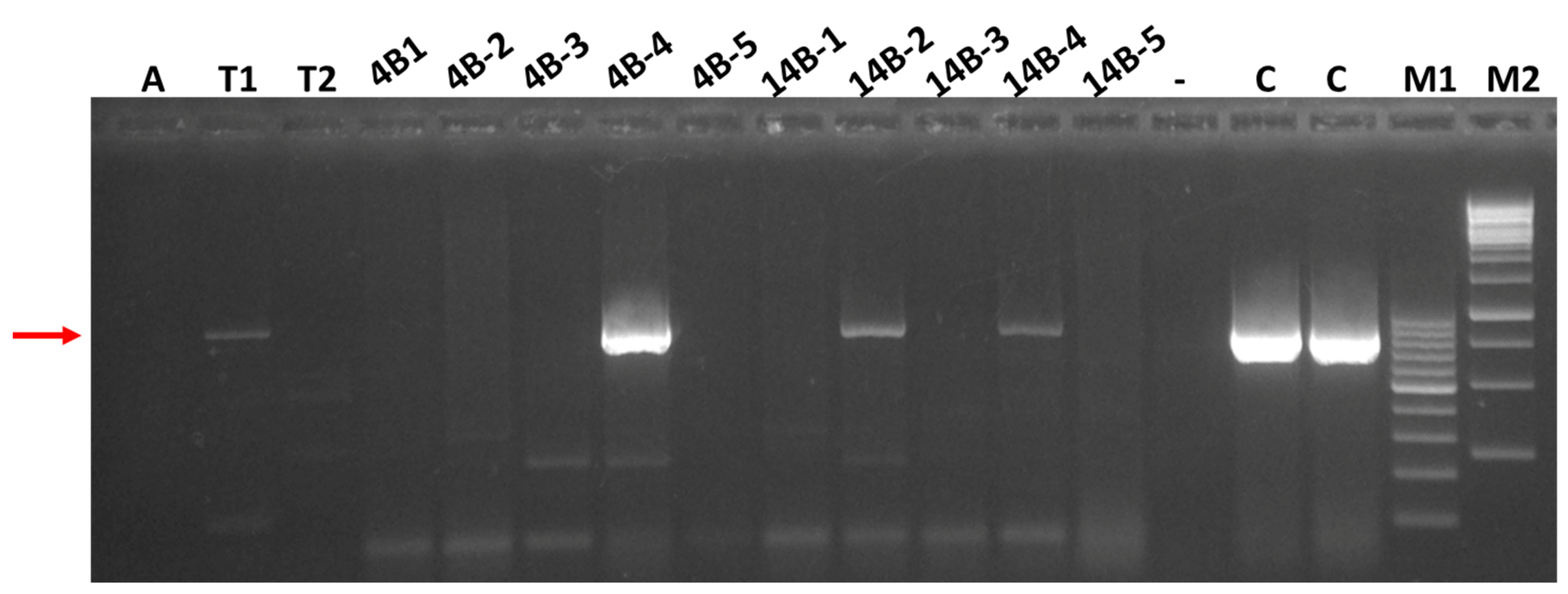
3.4. Attempts to Transmit SRAV and Detection of the Virus in Thrips Collected in Alfalfa Stands
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- U.S. Department of Agriculture-National Agricultural Statistics Service (USDA-NASS). 2021 State Agriculture Overview: Idaho. 2021. Available online: https://www.nass.usda.gov/Quick_Stats/Ag_Overview/stateOverview.php?state=IDAHO (accessed on 7 May 2022).
- Larsen, R.C. Diseases caused by viruses. In Compendium of Alfalfa Diseases and Pests, 3rd ed.; Samac, D.A., Rhodes, L.H., Lamp, W.O., Eds.; The American Phytopathological Society: St. Paul, MN, USA, 2015; pp. 66–72. [Google Scholar]
- Thekke-Veetil, T.; Lagos-Kutz, D.; McCoppin, N.K.; Hartman, G.L.; Ju, H.-K.; Lim, H.-S.; Domier, L.L. Soybean Thrips (Thysanoptera: Thripidae) Harbor Highly Diverse Populations of Arthropod, Fungal and Plant Viruses. Viruses 2020, 12, 1376. [Google Scholar] [CrossRef] [PubMed]
- Nemchinov, L.G.; Irish, B.M.; Grinstead, S.; Shao, J.; Vieira, P. Diversity of the virome associated with alfalfa (Medicago sativa L.) in the U.S. Pacific Northwest. Sci. Rep. 2022, 12, 8726. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Roumagnac, P.; Nemchinov, L.G. High-throughput sequencing for deciphering the virome of alfalfa (Medicago sativa L.). Front. Microbiol. 2020, 11, 553109. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Debat, H.; Nome, C.; Cabrera-Mederos, D.; Trucco, V.; de Breuil, S.; Lenardon, S.; Giolitti, F. Redefining the Medicago sativa alphapartitiviruses genome sequences. Virus Res. 2019, 265, 156–161. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Richert-Pöggeler, K.R.; Maaß, C.; Vetten, H.-J.; Ziebell, H. Characterisation of a novel nucleorhabdovirus infecting alfalfa (Medicago sativa). Virol. J. 2019, 16, 55. [Google Scholar] [CrossRef] [Green Version]
- François, S.; Antoine-Lorquin, A.; Kulikowski, M.; Frayssinet, M.; Filloux, D.; Fernandez, E.; Roumagnac, P.; Froissart, R.; Ogliastro, M. Characterisation of the viral community associated with the alfalfa weevil (Hypera postica) and its host plant, alfalfa (Medicago sativa). Viruses 2021, 13, 791. [Google Scholar] [CrossRef]
- Jiang, P.; Shao, J.; Nemchinov, L.G. Identification of emerging viral genomes in transcriptomic datasets of alfalfa (Medicago sativa L.). Virol. J. 2019, 16, 153. [Google Scholar] [CrossRef]
- Nemchinov, L.G.; François, S.; Roumagnac, P.; Ogliastro, M.; Hammond, R.W.; Mollov, D.S.; Filloux, D. Characterization of alfalfa virus F, a new member of the genus Marafivirus. PLoS ONE 2018, 13, e0203477. [Google Scholar] [CrossRef]
- Nemchinov, L.G.; Grinstead, S.C.; Mollov, D.S. Alfalfa virus S, a new species in the family Alphaflexiviridae. PLoS ONE 2017, 12, e0178222. [Google Scholar] [CrossRef] [Green Version]
- Nemchinov, L.G.; Shao, J.; Postnikova, O.A. Complete genome sequence of the Alfalfa latent virus. Genome Announc. 2015, 3, e00250-15. [Google Scholar] [CrossRef] [Green Version]
- Samarfard, S.; McTaggart, A.R.; Sharman, M.; Bejerman, N.E.; Dietzgen, R.G. Viromes of ten alfalfa plants in Australia reveal diverse known viruses and a novel RNA virus. Pathogens 2020, 9, 214. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Park, D.; Hahn, Y. Identification of novel RNA viruses in alfalfa (Medicago sativa): An Alphapartitivirus, a Deltapartitivirus, and a Marafivirus. Gene 2018, 638, 7–12. [Google Scholar] [CrossRef]
- Nibert, M.L.; Pyle, J.D.; Firth, A.E. A +1 ribosomal frameshifting motif prevalent among plant amalgaviruses. Virology 2016, 498, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Tzanetakis, I.E.; Sabanadzovic, S.; Valverde, R.A. Amalgaviruses (Amalgaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D., Zuckerman, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 3. [Google Scholar]
- Li, J.; Gu, H.; Liu, Y.; Wei, S.; Hu, G.; Wang, X.; McNeill, M.R.; Ban, L. RNA-seq reveals plant virus composition and diversity in alfalfa, thrips, and aphids in Beijing, China. Arch. Virol. 2021, 166, 1711–1722. [Google Scholar] [CrossRef]
- Nemchinov, L.G.; Grinstead, S.; Irish, B.M.; Shao, J. Identification and complete genome sequencing of Alfalfa Virus S diagnosed in alfalfa plants (Medicago sativa) from Washington state, USA. Plant Dis. 2020, 104, 3271. [Google Scholar] [CrossRef]
- Feng, X.; Poplawsky, A.R.; Nikolaeva, O.V.; Myers, J.R.; Karasev, A.V. Recombinants of Bean common mosaic virus (BCMV) and Genetic Determinants of BCMV Involved in Overcoming Resistance in Common Bean. Phytopathology 2014, 104, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Green, K.J.; Mollov, D.; Tran, L.T.; Alvarez-Quinto, R.A.; Ochoa, J.B.; Quito-Avila, D.F.; Karasev, A.V. Characterization of a new tymovirus causing stunting and chlorotic mosaic in naranjilla (Solanum quitoense). Plant Dis. 2018, 102, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef] [Green Version]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Green, K.J.; Brown, C.J.; Gray, S.M.; Karasev, A.V. Phylogenetic study of recombinant strains of potato virus Y. Virology 2017, 507, 40–52. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE v5 Enables Improved Estimates of Phylogenetic Tree Confidence by Ensemble Bootstrapping. bioRxiv 2021. [Google Scholar] [CrossRef]
- Esterman, E.S.; Wolf, Y.I.; Kogay, R.; Koonin, E.V.; Zhaxybayeva, O. Evolution of DNA packaging in gene transfer agents. Virus Evol. 2021, 7, veab015. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessomoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Neri, U.; Wolf, Y.I.; Roux, S.; Camargo, A.P.; Lee, B.; Kazlauskas, D.; Chen, I.M.; Ivanova, N.; Allen, L.Z.; Paez-Espino, D.; et al. A Five-Fold Expansion of the Global RNA Virome Reveals Multiple New Clades of RNA Bacteriophages. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bratuleanu, B.E.; Temmam, S.; Chrétien, D.; Regnault, B.; Pérot, P.; Bouchier, C.; Bigot, T.; Savuța, G.; Eloit, M. The virome of Rhipicephalus, Dermacentor and Haemaphysalis ticks from Eastern Romania includes novel viruses with potential relevance for public health. Transbound. Emerg. Dis. 2021, 69, 1387–1403. [Google Scholar] [CrossRef]
- Fairman-Williams, M.E.; Guenther, U.-P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struc. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V. Computer-assisted identification of a putative methyltransferase domain in NS5 protein of flaviviruses and λ2 protein of reovirus. J. Gen. Virol. 1993, 74, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Blitvich, B.J.; Firth, A.E. Insect-Specific Flaviviruses: A Systematic Review of Their Discovery, Host Range, Mode of Transmission, Superinfection Exclusion Potential and Genomic Organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef] [Green Version]
- Colmant, A.M.G.; Hobson-Peters, J.; Bielefeldt-Ohmann, H.; van den Hurk, A.F.; Hall-Mendelin, S.; Chow, W.K.; Johansen, C.A.; Fros, J.; Simmonds, P.; Watterson, D.; et al. A new clade of insect-specific flaviviruses from Australian Anopheles mosquitoes displays species-specific host restriction. mSphere 2017, 2, e00262-17. [Google Scholar] [CrossRef] [Green Version]
- Bekal, S.; Domier, L.L.; Niblack, T.L.; Lambert, K.N.Y. Discovery and initial analysis of novel viral genomes in the soybean cyst nematode. J. Gen. Virol. 2011, 92, 1870–1879. [Google Scholar] [CrossRef]
- Kobayashi, K.; Atsumi, G.; Iwadate, Y.; Tomita, R.; Chiba, K.; Akasaka, S.; Nishihara, M.; Takahashi, H.; Yamaoka, N.; Nishiguchi, M.; et al. Gentian Kobu-sho-associated virus: A tentative, novel double-stranded RNA virus that is relevant to gentian Kobu-sho syndrome. J. Gen. Plant Pathol. 2013, 79, 56–63. [Google Scholar] [CrossRef]
- Atsumi, G.; Tomita, R.; Kobayashi, K.; Sekine, K.-T. Prevalence and genetic diversity of an unusual virus associated with Kobu-sho disease of gentian in Japan. J. Gen. Virol. 2013, 94, 2360–2365. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location/Sample Type | Collected Specimens | Time of Collection | Number of Samples | Method of Analysis |
|---|---|---|---|---|
| Location 1: Minidoka (ID), leaf tissue | Alfalfa | July 2020 | 6 | HTS, RT-PCR |
| Location 2: Twin Falls (ID), leaf tissue | Alfalfa | July 2021 | 25 | RT-PCR |
| Location 2: Twin Falls (ID), leaf tissue | Alfalfa | August 2021 | 10 | RT-PCR |
| Location 2: Twin Falls (ID), insects | Thrips | August 2021 | 2 | RT-PCR |
| Aphids | August 2021 | 1 | RT-PCR |
| Viruses (a) | ALF1059 | ALF1060 | ALF1061 | ALF1067 | ALF1071 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Contig Size, nt (b) | Mapped Reads, # (c) | Contig Size, nt | Mapped Reads, # | Contig Size, nt | Mapped Reads, # | Contig Size, nt | Mapped Reads, # | Contig Size, nt | Mapped Reads, # | |
| AMV | ||||||||||
| RNA1 | 3620 | 952,345 | 3627 | 559,215 | 3629 | 859,660 | 3630 | 680,142 | 3541 | 1224,303 |
| RNA2 | 1867 | 361,173 | 2497 | 169,385 | 2575 | 466,668 | 2563 | 559,374 | 2572 | 512,950 |
| RNA3 | 2029 | 507,736 | 2012 | 182,108 | 2010 | 732,928 | 1999 | 353,662 | 2017 | 750,965 |
| BLRV | - | 2 | - | 4 | - | 12 | 4162 | 5895 | 5895 | 3207 |
| MsAPV1 | ||||||||||
| RNA1 | 1903 | 5378 | 1996 | 10,463 | 1870 | 11,193 | 1774 | 14,650 | 1633 | 22,723 |
| RNA2 | 1790 | 422 | 1679 | 725 | 1793 | 551 | 1785 | 688 | 1757 | 1001 |
| MsAPV2 | ||||||||||
| RNA1 | - | 0 | 1902 | 1236 | - | 2 | - | 0 | - | 0 |
| RNA2 | - | 0 | 1771 | 161 | - | 0 | - | 0 | - | 0 |
| MsDPV1 | ||||||||||
| RNA1 | - | 2 | - | 0 | - | 20 | - | 0 | 1552 | 45 |
| RNA2 | 496 | 4 | - | 0 | 1503 | 57 | - | 0 | 1353 | 134 |
| MsAV1 | 2905 | 154 | 3369 | 177 | 3386 | 293 | 3415 | 193 | - | 0 |
| SRAV | - | 0 | 11,803 | 1269 | 7788 | 275 | - | 0 | 11,797 | 1662 |
| Collection Date/Field | Planting Year | AMV-R2 | BLRV | MsAPV1-R1 | MsAPV2-R1 | MsDPV-R1 | MsAV1 | SRAV |
|---|---|---|---|---|---|---|---|---|
| July 2020, Rupert, ID | ||||||||
| NE | 2017 | 6/6 | 4/6 | 6/6 | 2/6 | 3/6 | 5/6 | 4/6 |
| July 2021, Kimberly, ID | ||||||||
| 4B | 2020 | 0/5 | 2/5 | 5/5 | 2/5 | 3/5 | 2/5 | 3/5 |
| 14B | 2020 | 0/5 | 0/5 | 3/5 | 1/5 | 4/5 | 3/5 | 2/5 |
| 23S | 2019 | 1/5 | 3/5 | 5/5 | 1/5 | 1/5 | 2/5 | 3/5 |
| 7C | 2017 | 5/5 | 4/5 | 5/5 | 2/5 | 1/5 | 2/5 | 4/5 |
| 59A | 2013 | 3/5 | 5/5 | 5/5 | 1/5 | 0/5 | 1/5 | 3/5 |
| August 2021, Kimberly, ID | ||||||||
| 4B | 2020 | 5/5 | 5/5 | 5/5 | 3/5 | 3/5 | 3/5 | 1/5 |
| 14B | 2020 | 3/5 | 5/5 | 5/5 | 2/5 | 1/5 | 4/5 | 2/5 |
| Sample Origin (Field and ID) | 5 wpi | 8 wpi | ||
|---|---|---|---|---|
| AMV | SRAV | AMV | SRAV | |
| 4B-1 | 3/3 | 0/3 | 3/3 | 0/3 |
| 4B-2 | 2/3 | 0/3 | 3/3 | 0/3 |
| 4B-3 | 0/3 | 0/3 | 0/3 | 0/3 |
| 4B-4 | 3/3 | 0/3 | 3/3 | 0/3 |
| 4B-5 | 3/3 | 0/3 | 3/3 | 0/3 |
| 14B-1 | 3/3 | 0/3 | 3/3 | 0/3 |
| 14B-2 | 0/3 | 0/3 | 3/3 | 0/3 |
| 14B-3 | 0/3 | 0/3 | 3/3 | 0/3 |
| 14B-4 | 0/3 | 0/3 | 3/3 | 0/3 |
| 14B-5 | 3/3 | 0/3 | 3/3 | 0/3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahan, J.; Wolf, Y.I.; Orellana, G.E.; Wenninger, E.J.; Koonin, E.V.; Karasev, A.V. A Novel Flavi-like Virus in Alfalfa (Medicago sativa L.) Crops along the Snake River Valley. Viruses 2022, 14, 1320. https://doi.org/10.3390/v14061320
Dahan J, Wolf YI, Orellana GE, Wenninger EJ, Koonin EV, Karasev AV. A Novel Flavi-like Virus in Alfalfa (Medicago sativa L.) Crops along the Snake River Valley. Viruses. 2022; 14(6):1320. https://doi.org/10.3390/v14061320
Chicago/Turabian StyleDahan, Jennifer, Yuri I. Wolf, Gardenia E. Orellana, Erik J. Wenninger, Eugene V. Koonin, and Alexander V. Karasev. 2022. "A Novel Flavi-like Virus in Alfalfa (Medicago sativa L.) Crops along the Snake River Valley" Viruses 14, no. 6: 1320. https://doi.org/10.3390/v14061320
APA StyleDahan, J., Wolf, Y. I., Orellana, G. E., Wenninger, E. J., Koonin, E. V., & Karasev, A. V. (2022). A Novel Flavi-like Virus in Alfalfa (Medicago sativa L.) Crops along the Snake River Valley. Viruses, 14(6), 1320. https://doi.org/10.3390/v14061320