
Selection and Validation of Reference Genes for Gene Expression Studies Using Quantitative Real-Time PCR in Prunus Necrotic Ringspot Virus-Infected Cucumis sativus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Virus Inoculation
2.2. Total RNA Isolation and First-Strand cDNA Synthesis
2.3. Selection of Candidate Reference Genes and Primer Design
2.4. RT-qPCR Analysis
2.5. Data Analyses for Expression Stability
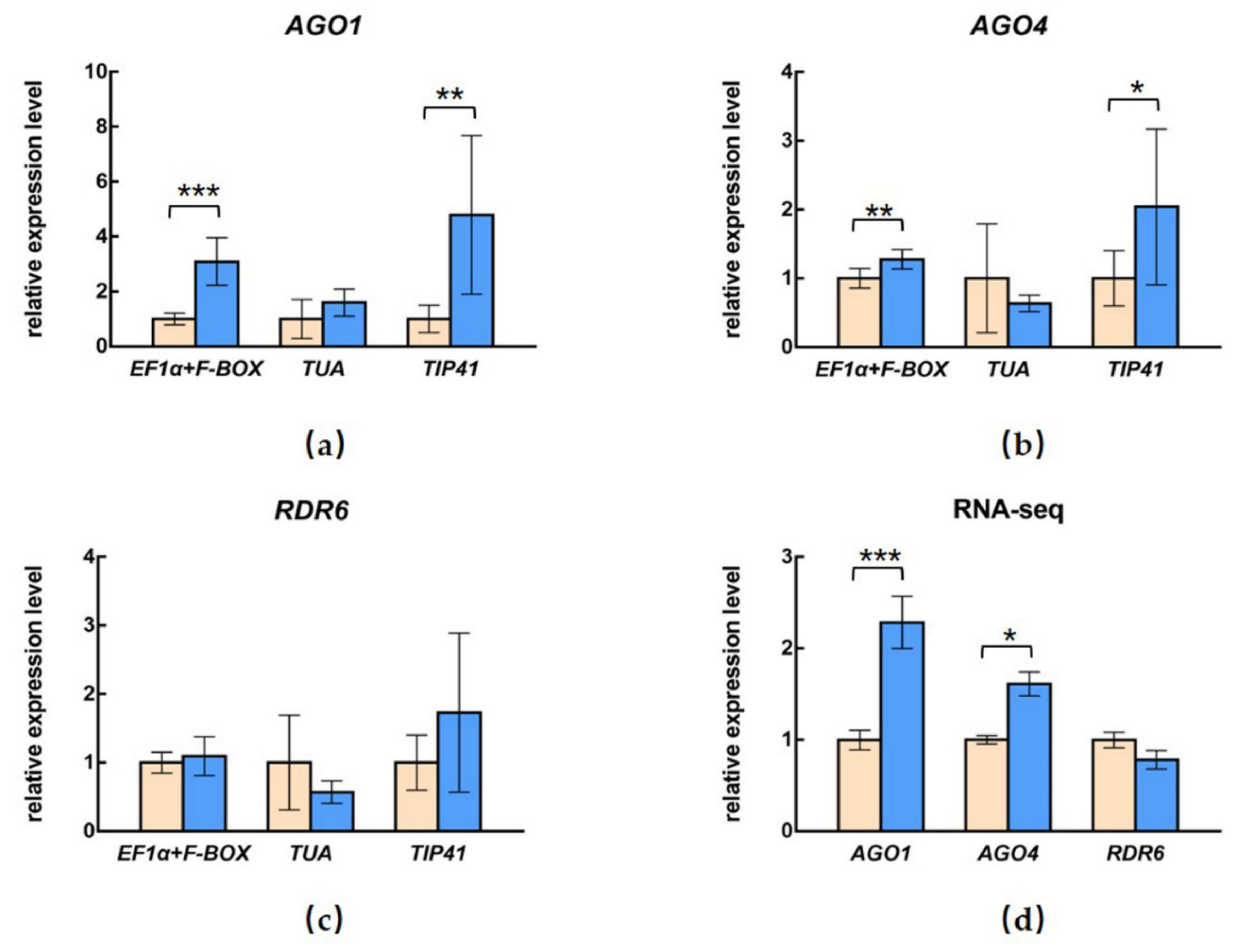
2.6. Relative Quantification of AGO1, AGO4, and RDR6
2.7. High-Throughput RNA-Sequencing and Analysis of the Sequence Data
3. Results
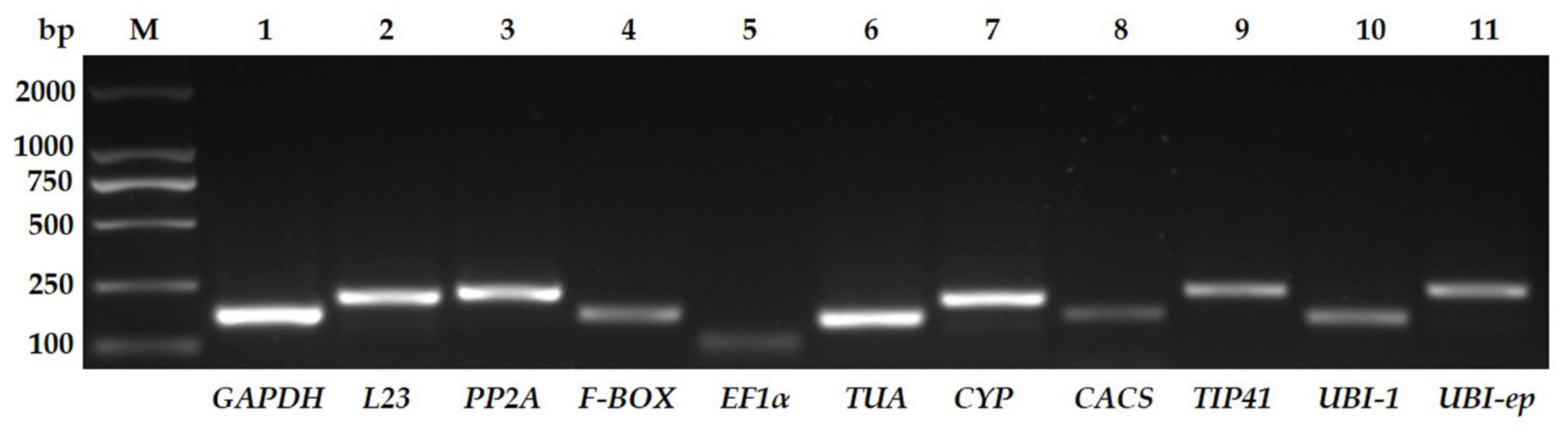
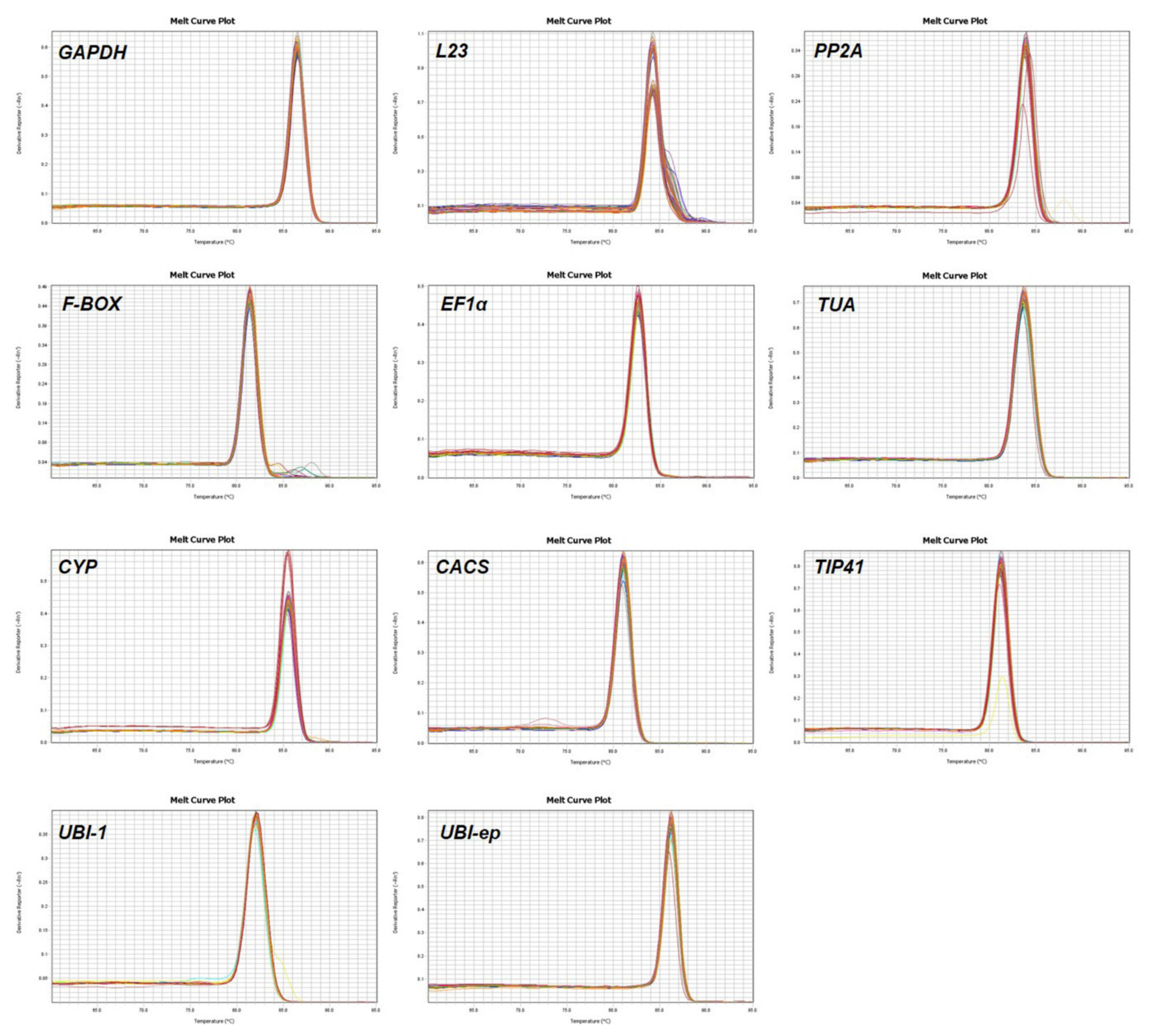
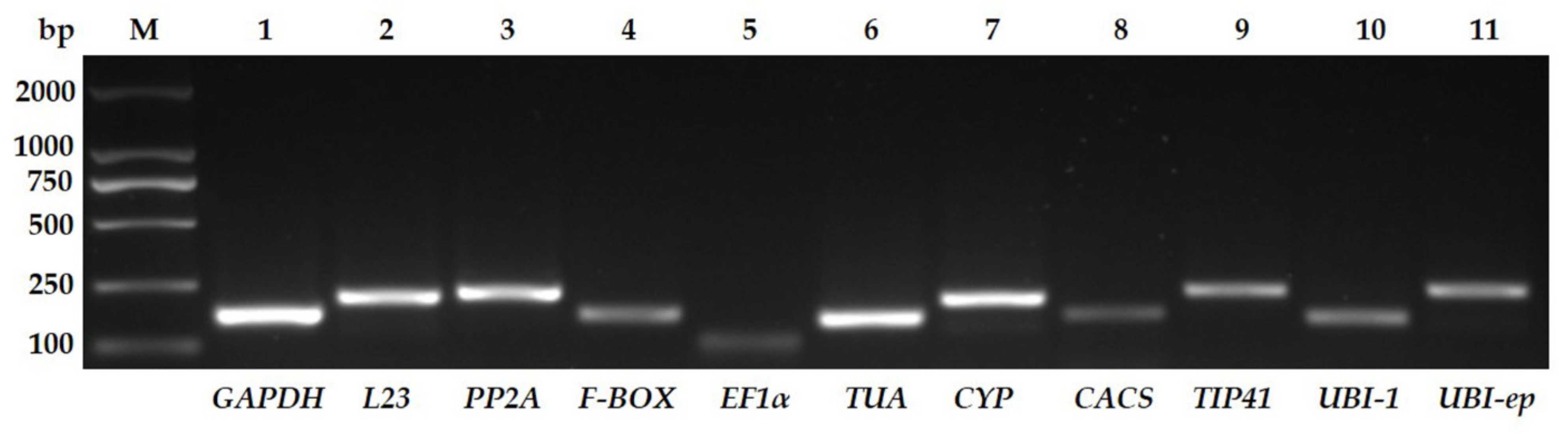
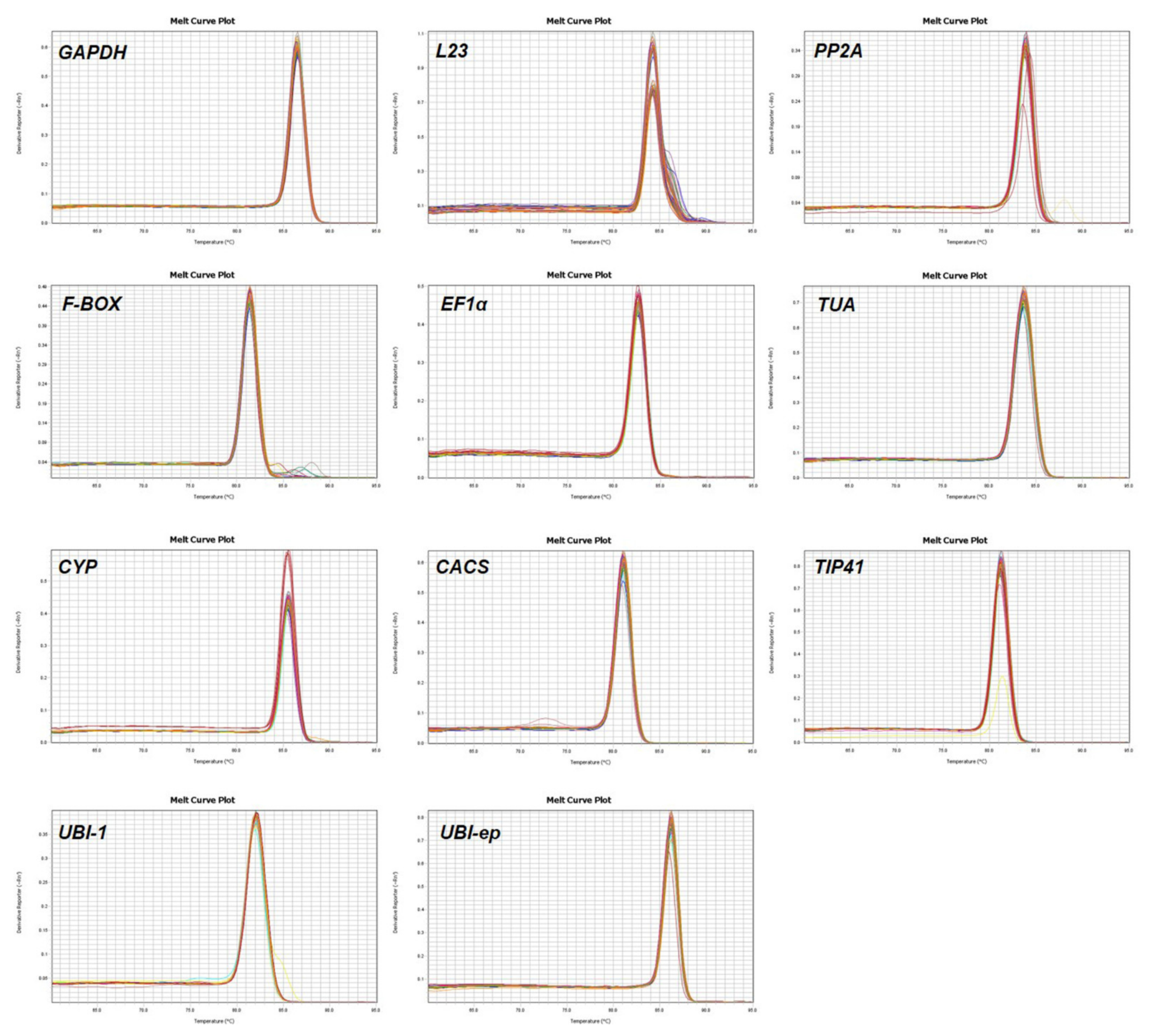
3.1. Assessment of Primer Specificity and Amplification Efficiency
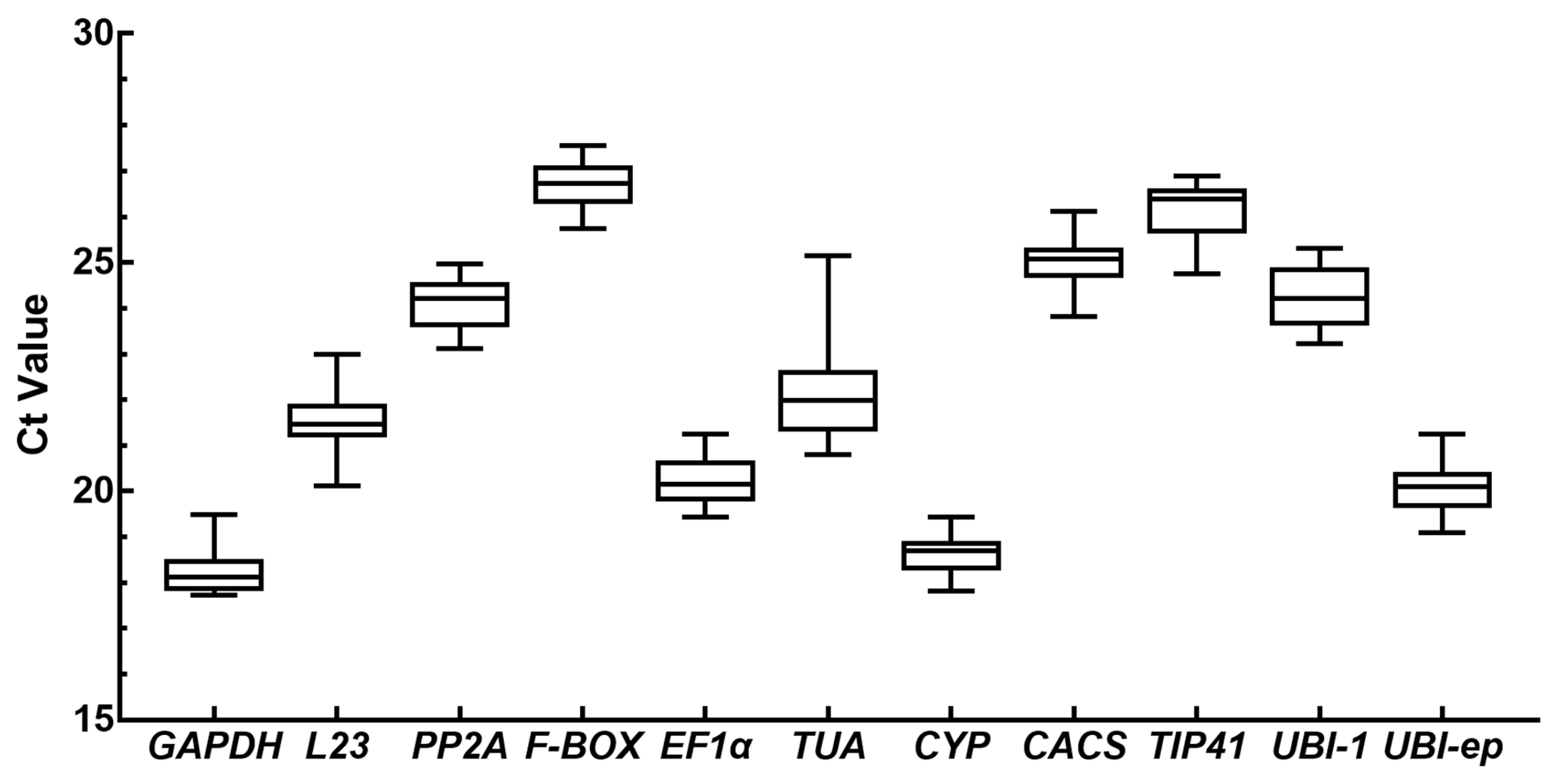
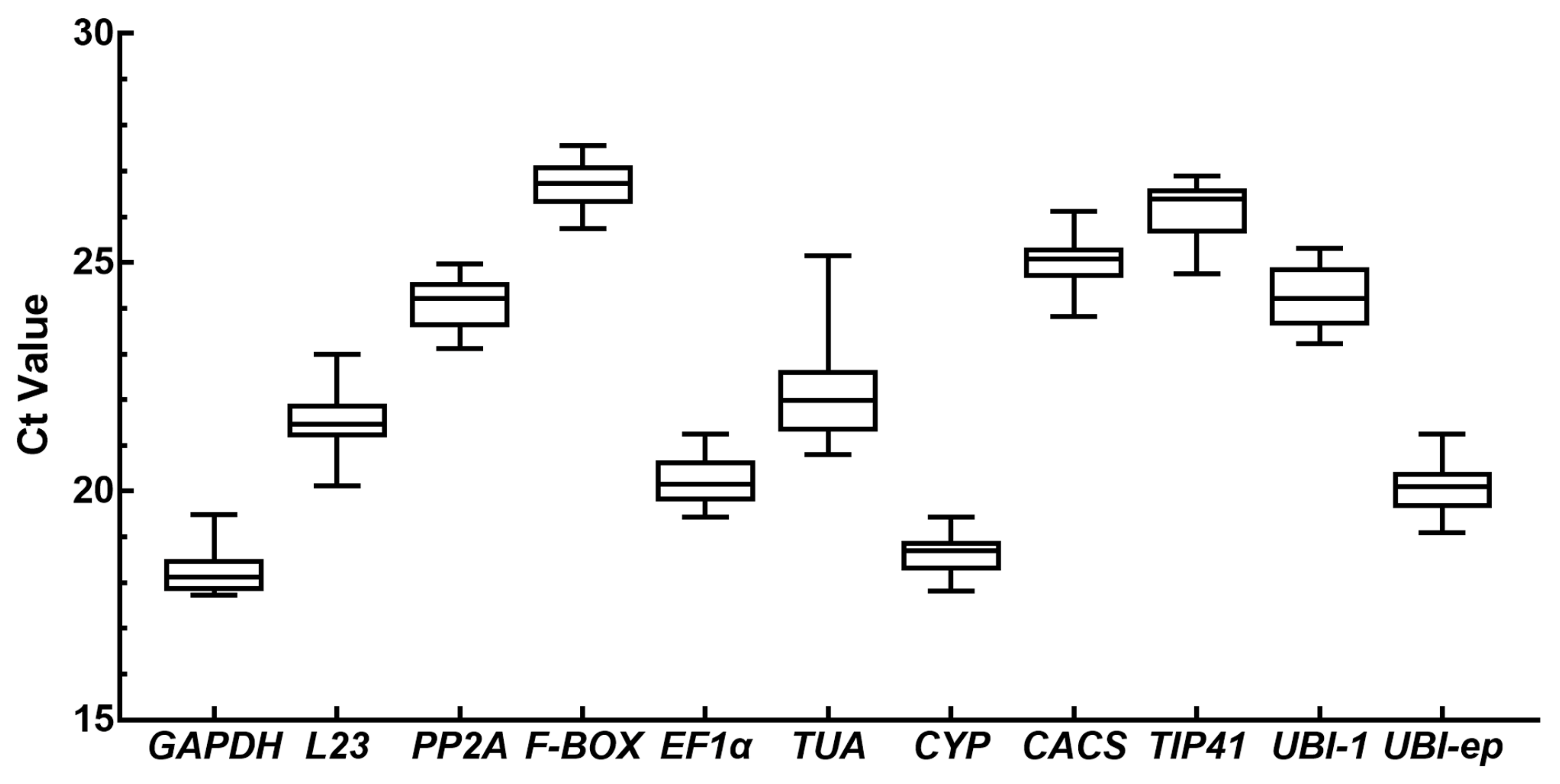
3.2. Expression Levels and Variation in Candidate Reference Genes
3.3. Analyses of Candidate Reference Gene Stability Using Four Different Types of Software
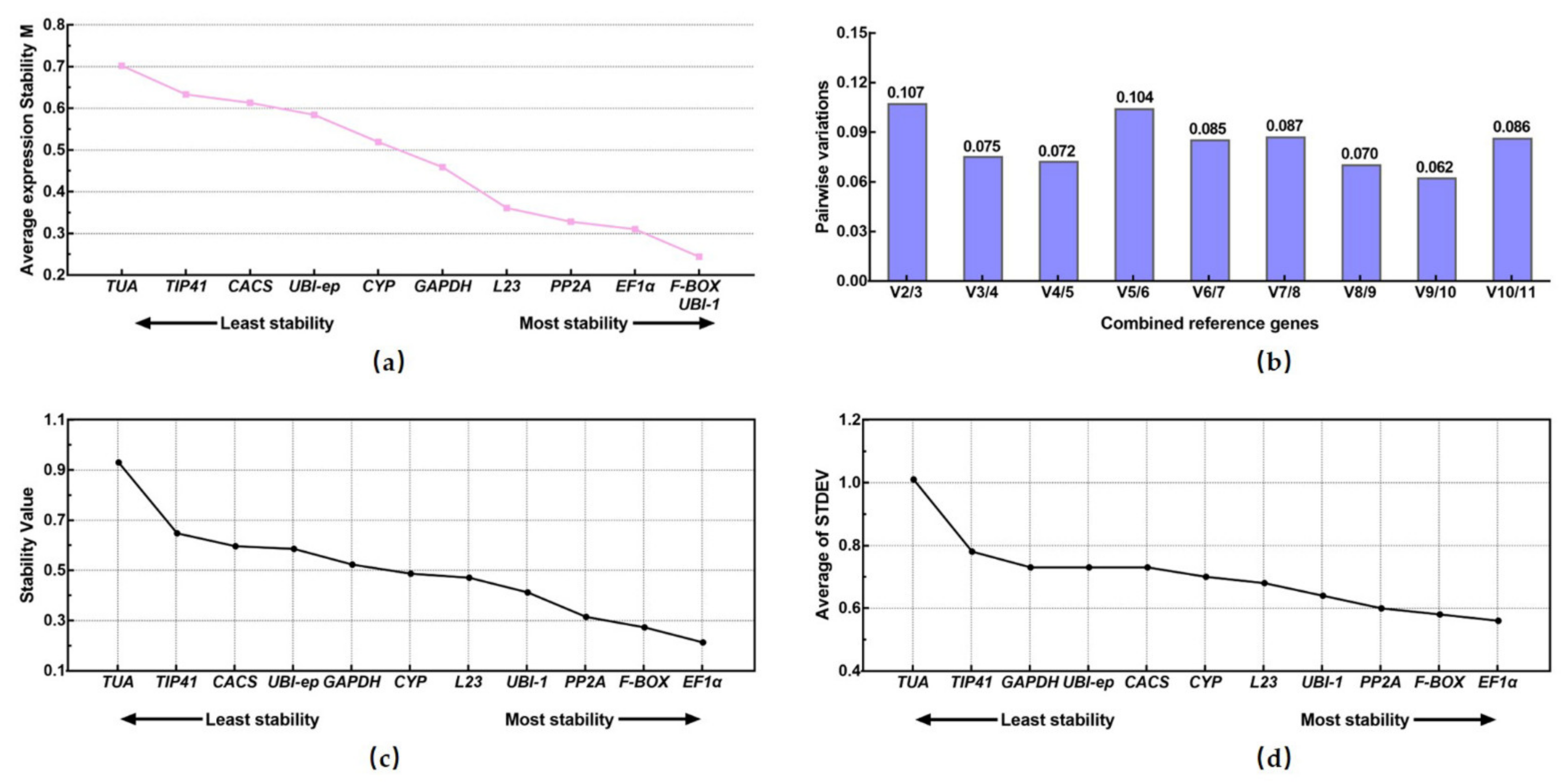
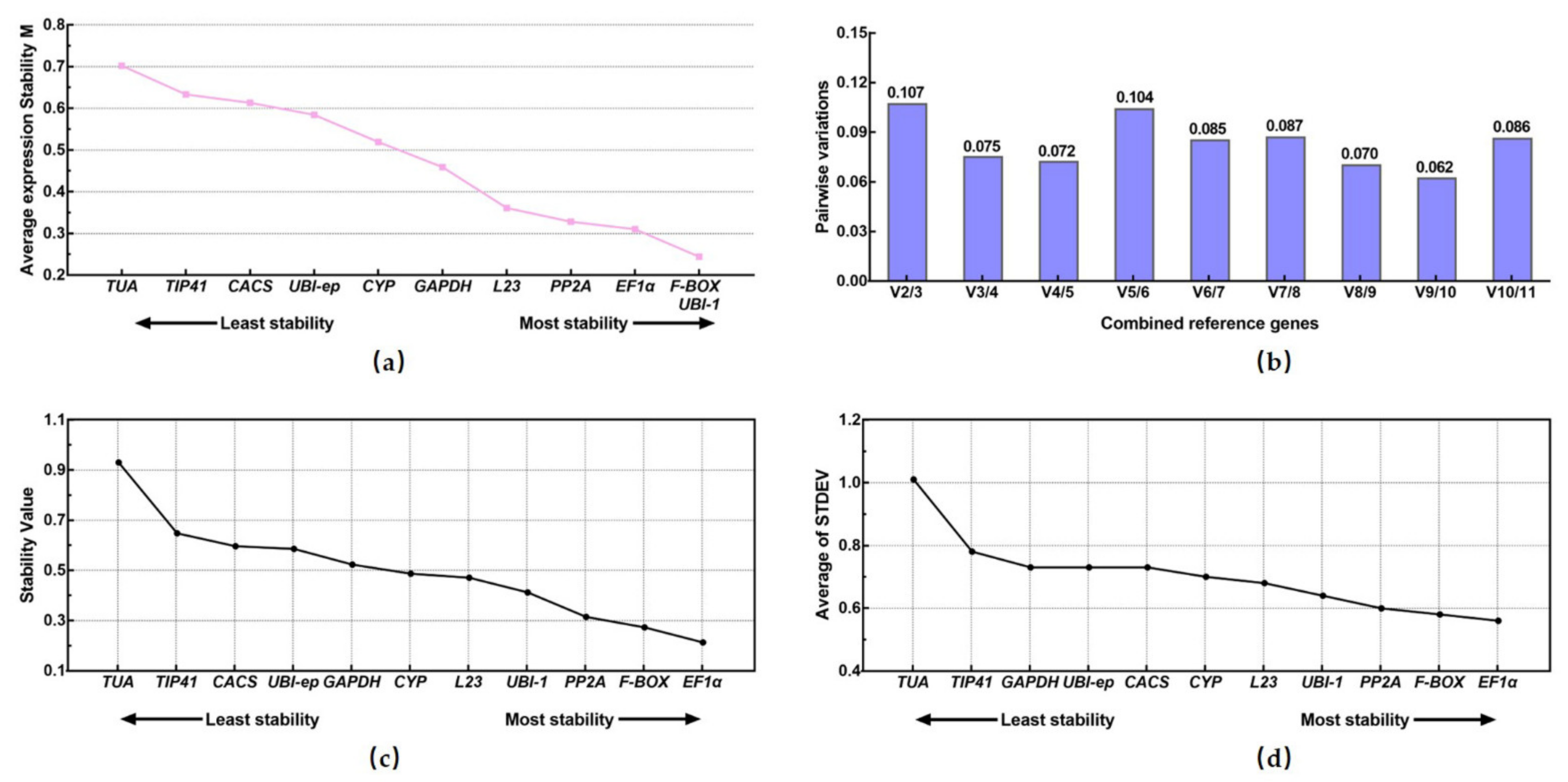
3.3.1. geNorm Analysis
3.3.2. NormFinder Analysis
3.3.3. Δ.Ct Analysis
3.3.4. Bestkeeper Analysis
3.3.5. RefFinder Analysis
3.4. Influence of Different Selection of Reference Genes on the Relative Expression of Target Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kozian, D.H.; Kirschbaum, B.J. Comparative gene-expression analysis. Trends Biotechnol. 1999, 17, 73–78. [Google Scholar] [CrossRef]
- Robert, C. Microarray analysis of gene expression during early development: A cautionary overview. Reproduction 2010, 140, 787–801. [Google Scholar] [CrossRef] [PubMed]
- De Klerk, E.; AC’t Hoen, P. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef]
- Allison, D.B.; Cui, X.; Page, G.P.; Sabripour, M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006, 7, 55–65. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Guenin, S.; Mauriat, M.; Pelloux, J.; Van Wuytswinkel, O.; Bellini, C.; Gutierrez, L. Normalization of qRT-PCR data: The necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot. 2009, 60, 487–493. [Google Scholar] [CrossRef]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.-R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef]
- Sundaram, V.K.; Sampathkumar, N.K.; Massaad, C.; Grenier, J. Optimal use of statistical methods to validate reference gene stability in longitudinal studies. PLoS ONE 2019, 14, e0219440. [Google Scholar] [CrossRef]
- Lilly, S.T.; Drummond, R.S.; Pearson, M.N.; MacDiarmid, R.M. Identification and validation of reference genes for normalization of transcripts from virus-infected Arabidopsis thaliana. Mol. Plant-Microbe Interact. 2011, 24, 294–304. [Google Scholar] [CrossRef]
- Liu, D.; Shi, L.; Han, C.; Yu, J.; Li, D.; Zhang, Y. Validation of reference genes for gene expression studies in virus-infected Nicotiana benthamiana using quantitative real-time PCR. PLoS ONE 2012, 7, e46451. [Google Scholar] [CrossRef]
- Petit, C.; Pernin, F.; Heydel, J.-M.; Délye, C. Validation of a set of reference genes to study response to herbicide stress in grasses. BMC Res. Notes 2012, 5, 18. [Google Scholar] [CrossRef]
- Baek, E.; Yoon, J.-Y.; Palukaitis, P. Validation of reference genes for quantifying changes in gene expression in virus-infected tobacco. Virology 2017, 510, 29–39. [Google Scholar] [CrossRef]
- Mascia, T.; Santovito, E.; Gallitelli, D.; Cillo, F. Evaluation of reference genes for quantitative reverse-transcription polymerase chain reaction normalization in infected tomato plants. Mol. Plant Pathol. 2010, 11, 805–816. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Pihur, V.; Datta, S.; Datta, S. RankAggreg, an R package for weighted rank aggregation. BMC Bioinf. 2009, 10, 62. [Google Scholar] [CrossRef]
- Schmidt, G.W.; Delaney, S.K. Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol. Genet. Genomics 2010, 283, 233–241. [Google Scholar] [CrossRef]
- An, J.; Shen, X.; Ma, Q.; Yang, C.; Liu, S.; Chen, Y. Transcriptome profiling to discover putative genes associated with paraquat resistance in goosegrass (Eleusine indica L.). PLoS ONE 2014, 9, e99940. [Google Scholar] [CrossRef]
- Moretti, M.L.; Alárcon-Reverte, R.; Pearce, S.; Morran, S.; Hanson, B.D. Transcription of putative tonoplast transporters in response to glyphosate and paraquat stress in Conyza bonariensis and Conyza canadensis and selection of reference genes for qRT-PCR. PLoS ONE 2017, 12, e0180794. [Google Scholar] [CrossRef]
- Reddy, P.S.; Reddy, D.S.; Sivasakthi, K.; Bhatnagar-Mathur, P.; Vadez, V.; Sharma, K.K. Evaluation of sorghum [Sorghum bicolor (L.)] reference genes in various tissues and under abiotic stress conditions for quantitative real-time PCR data normalization. Front. Plant Sci. 2016, 7, 529. [Google Scholar] [CrossRef]
- Zhu, H.; Ma, Y.; Guo, Q. Expression stability of internal reference gene in response to Trichoderma polysporum infection in Avena fatua L. Curr. Genet. 2021, 67, 909–918. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, X.; Chen, S.; Zheng, L.; He, X.; Liu, M.; Qiao, G.; Wang, Y.; Zhuo, R. Selection of suitable reference genes for quantitative real-time PCR gene expression analysis in Salix matsudana under different abiotic stresses. Sci. Rep. 2017, 7, 40290. [Google Scholar] [CrossRef]
- Lee, J.M.; Roche, J.R.; Donaghy, D.J.; Thrush, A.; Sathish, P. Validation of reference genes for quantitative RT-PCR studies of gene expression in perennial ryegrass (Lolium perenne L.). BMC Mol. Biol. 2010, 11, 8. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, Z.; Qin, L.; Zhou, Y.; Fan, H.; Zhang, Z.; Wu, Z.; Li, S. Incidence of major peach viruses and viroids in China. J. Plant Pathol. 2013, 95, 603–607. [Google Scholar]
- Manganaris, G.A.; Economou, A.S.; Boubourakas, I.N.; Katis, N.I. Elimination of PPV and PNRSV through thermotherapy and meristem-tip culture in nectarine. Plant Cell Rep. 2003, 22, 195–200. [Google Scholar] [CrossRef]
- Mekuria, G.; Ramesh, S.A.; Alberts, E.; Bertozzi, T.; Wirthensohn, M.; Collins, G.; Sedgley, M. Comparison of ELISA and RT-PCR for the detection of prunus necrotic ring spot virus and prune dwarf virus in almond (Prunus dulcis). J. Virol. Methods 2003, 114, 65–69. [Google Scholar] [CrossRef]
- Zong, X.J.; Liu, Q.Z.; Wang, W.W.; Wei, H.R.; Wang, J.W. Detection of prunus necrotic ringspot virus and prune dwarf virus in sweet cherry (Prunus avium) and Chinese cherry (Prunus pseudocerasus) in Shandong Province, China. In Proceedings of the VII International Cherry Symposium, Plasencia, Spain, 23 June 2013; pp. 469–474. [Google Scholar]
- Oliver, J.E.; Freer, J.; Andersen, R.L.; Cox, K.D.; Robinson, T.L.; Fuchs, M. Genetic diversity of prunus necrotic ringspot virus isolates within a cherry orchard in New York. Plant Dis. 2009, 93, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Kinoti, W.M.; Nancarrow, N.; Dann, A.; Rodoni, B.C.; Constable, F.E. Updating the quarantine status of prunus infecting viruses in Australia. Viruses 2020, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Amari, K.; Diaz-Vivancos, P.; Pallas, V.; Amelia Sanchez-Pina, M.; Antonio Hernandez, J. Oxidative stress induction by prunus necrotic ringspot virus infection in apricot seeds. Physiol. Plant. 2007, 131, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.J.; Dong, Y.F.; Zhang, Z.P.; Fan, X.D.; Ren, F.; Li, Z.N.; Zhou, J. First report of prunus necrotic ringspot virus infection of apple in China. Plant Dis. 2016, 100, 1955–1956. [Google Scholar] [CrossRef]
- Xing, F.; Gao, D.; Liu, H.; Wang, H.; Habili, N.; Li, S. Molecular characterization and pathogenicity analysis of prunus necrotic ringspot virus isolates from China rose (Rosa chinensis Jacq.). Arch. Virol. 2020, 165, 2479–2486. [Google Scholar] [CrossRef]
- Cui, H.; Hong, N.; Wang, G.; Wang, A. Genomic segments RNA1 and RNA2 of prunus necrotic ringspot virus codetermine viral pathogenicity to adapt to alternating natural prunus hosts. Mol. Plant-Microbe Interact. 2013, 26, 515–527. [Google Scholar] [CrossRef]
- Pethybridge, S.J.; Wilson, C.R.; Ferrandino, F.J.; Leggett, G.W. Spatial analyses of viral epidemics in Australian hop gardens: Implications for mechanisms of spread. Plant Dis. 2000, 84, 513–515. [Google Scholar] [CrossRef]
- Pallas, V.; Aparicio, F.; Herranz, M.C.; Amari, K.; Sanchez-Pina, M.A.; Myrta, A.; Sanchez-Navarro, J.A. Ilarviruses of Prunus spp.: A continued concern for fruit trees. Phytopathology 2012, 102, 1108–1120. [Google Scholar] [CrossRef]
- Pappi, P.G.; Fotiou, I.; Efthimiou, K.E.; Katis, N.I.; Maliogka, V.I. Development of three duplex real-time RT-PCR assays for the sensitive and rapid detection of a phytoplasma and five viral pathogens affecting stone fruit trees. Mol. Cell. Probes 2020, 53, 101621. [Google Scholar] [CrossRef]
- Sharma, A.; Ram, R.; Zaidi, A.A. Rubus ellipticus, a perennial weed host of prunus necrotic ring spot virus in India. Plant Dis. 1998, 82, 1283. [Google Scholar] [CrossRef]
- Akbaş, B.; Ilhan, H.D. Widespread distribution of apple mosaic virus on apple in Turkey. Plant Dis. 2005, 89, 1010. [Google Scholar] [CrossRef]
- Kinoti, W.; Plummer, K.; Constable, F.E.; Nancarrow, N.; Rodoni, B. The partial characterization of ilarviruses infecting prunus species in Australia. In Proceedings of the XXIX International Horticultural Congress on Horticulture: Sustaining Lives, Livelihoods and Landscapes (IHC2014): International Symposium on Nut Crops, Brisbane, QL, Australia, 23 February 2016; pp. 243–247. [Google Scholar]
- Liu, M.; Liang, Z.; Aranda, M.A.; Hong, N.; Liu, L.; Kang, B.; Gu, Q. A cucumber green mottle mosaic virus vector for virus-induced gene silencing in cucurbit plants. Plant Methods 2020, 16, 9. [Google Scholar] [CrossRef]
- Fang, L.; Wei, X.; Liu, L.; Zhou, L.; Tian, Y.; Geng, C.; Li, X. A tobacco ringspot virus-based vector system for gene and microRNA function studies in cucurbits. Plant Physiol. 2021, 186, 853–864. [Google Scholar] [CrossRef]
- Wang, S.; Wang, R.; Liang, D.; Ma, F.; Shu, H. Molecular characterization and expression analysis of a glycine-rich RNA-binding protein gene from Malus hupehensis Rehd. Mol. Biol. Rep. 2012, 39, 4145–4153. [Google Scholar] [CrossRef]
- Wan, H.; Zhao, Z.; Qian, C.; Sui, Y.; Malik, A.A.; Chen, J. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 2010, 399, 257–261. [Google Scholar] [CrossRef]
- Warzybok, A.; Migocka, M. Reliable reference genes for normalization of gene expression in cucumber grown under different nitrogen nutrition. PLoS ONE 2013, 8, e72887. [Google Scholar] [CrossRef]
- Holmes, A.; Birse, L.; Jackson, R.W.; Holden, N.J. An optimized method for the extraction of bacterial mRNA from plant roots infected with Escherichia coli O157:H7. Front. Microbiol. 2014, 5, 286. [Google Scholar] [CrossRef]
- Chandel, V.; Rana, T.; Hallan, V.; Zaidi, A.A. Evidence for the occurrence of prunus necrotic ringspot virus on peach in India by serological and molecular methods. Can. J. Plant Pathol. 2007, 29, 311–316. [Google Scholar] [CrossRef]
- Sipahioglu, H.M.; Baloglu, S. The incidence of prunus necrotic ringspot (PNRSV) and apple chlorotic leafspot (ACLSV) viruses on stone fruits grown in East Anatolia region of Turkey. In Proceedings of the XII ISHS Symposium on Apricot Culture and Decline, Avignon, France, 28 February 2006; pp. 469–472. [Google Scholar]
- Noda, H.; Yamagishi, N.; Yaegashi, H.; Xing, F.; Xie, J.; Li, S.; Zhou, T.; Ito, T.; Yoshikawa, N. Apple necrotic mosaic virus, a novel ilarvirus from mosaic-diseased apple trees in Japan and China. J. Gen. Plant Pathol. 2017, 83, 83–90. [Google Scholar] [CrossRef]
- Xing, F.; Robe, B.L.; Zhang, Z.; Wang, H.; Li, S. Genomic analysis, sequence diversity, and occurrence of apple necrotic mosaic virus, a novel ilarvirus associated with mosaic disease of apple trees in China. Plant Dis. 2018, 102, 1841–1847. [Google Scholar] [CrossRef]
- Kovalskaya, N.; Hammond, R.W. Molecular biology of viroid–host interactions and disease control strategies. Plant Sci. 2014, 228, 48–60. [Google Scholar] [CrossRef]
- Migocka, M.; Papierniak, A. Identification of suitable reference genes for studying gene expression in cucumber plants subjected to abiotic stress and growth regulators. Mol. Breed. 2011, 28, 343–357. [Google Scholar] [CrossRef]
- Liang, C.; Hao, J.; Meng, Y.; Luo, L.; Li, J. Identifying optimal reference genes for the normalization of microRNA expression in cucumber under viral stress. PLoS ONE 2018, 13, e0194436. [Google Scholar] [CrossRef]
- Ding, S.-W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Full Name | Accession Number | Sequence (5′-3′) | Amplicon Length (bp) | Tm (°C) | E (%) | R2 | References |
|---|---|---|---|---|---|---|---|---|
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | NM_001305758 | F: CATCAAGGGAACAATGACTACCAC R: TAAGAGAAGGGAGGACCAAGGC | 161 | 58 | 95.21 | 0.9928 | This study |
| L23 | 60S ribosomal protein | XM_004140527 | F: AACAAGTTTCGGATGTCACTGG R: CAATAACAGCAGGCAAGACCTTC | 211 | 58 | 93.92 | 0.9946 | This study |
| PP2A | Protein phosphatase 2A | XM_004146712 | F: TGGAAGCACTTCACCGACCTCT R: CCAGCACCACGCGGAGATATTC | 224 | 58 | 94.37 | 0.9992 | This study |
| F-BOX | F-box protein | XM_031885920 | F: AAGCGTCTATTCCTTGTTGGTAGG R: CAAAGCACTCAAATCTCTCCGC | 166 | 58 | 96.24 | 0.9925 | This study |
| EF1α | Elongation factor 1-α | DQ341381 | F: ATTCAAGTATGCCTGGGTGC R: CAGTCAGCCTGTGATGTTCC | 174 | 58 | 92.10 | 0.9958 | Wan et al., 2012 [46] |
| TUA | α-Tubulin | AJ715498 | F: CATTCTCTCTTGGAACACACTGA R: TCAAACTGGCAGTTAAAGATGAAA | 106 | 58 | 95.74 | 0.9938 | Wan et al., 2010 [47] |
| CYP | Cyclophilin | AY942800 | F: GGAAATGGTACAGGAGGTG R: CATACCCTCAACGACTTGAC | 88 | 58 | 95.02 | 0.9984 | Wan et al., 2010 [47] |
| CACS | AP-2 complex subunit mu-1 | GT035008 | F: GTGCTTTCTTTCTGGAATGC R: TGAACCTCGTCAAATTTACACA | 158 | 58 | 93.12 | 0.9924 | Warzybok et al., 2013 [48] |
| TIP41 | TIP41-like family protein | GW881871 | F: CAACAGGTGATATTGGATTATGATTAC R: GCCAGCTCATCCTCATATAAG | 221 | 58 | 91.99 | 0.9928 | Warzybok et al., 2013 [48] |
| UBI-1 | Ubiquitin-like protein | AF104391 | F: CCAAAGCACAAGCAAGAGAC R: AGTAGGTTGTCTTATGGCGC | 143 | 58 | 93.29 | 0.9990 | Wan et al., 2010 [47] |
| UBI-ep | Ubiquitin extension protein | AY372537 | F: CACCAAGCCCAAGAAGATC R: TAAACCTAATCACCACCAGC | 220 | 58 | 92.93 | 0.9955 | Wan et al., 2010 [47] |
| AGO1 | Argonaute 1 | XM_011651337 | F: CGTTTCCCAGTGCTGTTTGAC R: GCCAATCTTGAGAAGCCACAAC | 245 | 58 | 94.79 | 0.9987 | This study |
| AGO4 | Argonaute 4 | XM_011655229 | F: GTCAAGCATCGGAACATCG R: TCCTTCTCAACGAATCGGAC | 223 | 58 | 94.73 | 0.9982 | This study |
| RDR6 | RNA dependent RNA polymerase 6 | XM_011650266 | F: TGTCTCACCTGACTCTGCTGCT R: ATTCCACTCGAAGGCCCTCTCC | 228 | 58 | 95.93 | 0.9961 | This study |
| Gene Name | geNorm | NormFinder | Delt Ct | BestKeeper | RefFinder | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| M-Value | Ranking | Stability | Ranking | Stability | Ranking | r Value | Ranking | Stability | Ranking | |
| EF1α | 0.310 | 3 | 0.213 | 1 | 0.56 | 1 | 0.873 | 1 | 1.73 | 1 |
| F-BOX | 0.244 | 1 | 0.273 | 2 | 0.58 | 2 | 0.826 | 4 | 2.00 | 2 |
| UBI-1 | 0.244 | 1 | 0.412 | 4 | 0.64 | 4 | 0.765 | 5 | 3.46 | 3 |
| CYP | 0.519 | 7 | 0.487 | 6 | 0.70 | 6 | 0.327 | 7 | 3.98 | 4 |
| PP2A | 0.328 | 4 | 0.315 | 3 | 0.60 | 3 | 0.845 | 3 | 4.12 | 5 |
| GAPDH | 0.459 | 6 | 0.523 | 7 | 0.73 | 7 | 0.387 | 8 | 4.92 | 6 |
| L23 | 0.361 | 5 | 0.471 | 5 | 0.68 | 5 | 0.761 | 6 | 5.44 | 7 |
| UBI-ep | 0.584 | 8 | 0.586 | 8 | 0.73 | 7 | 0.356 | 9 | 7.11 | 8 |
| CACS | 0.613 | 9 | 0.596 | 9 | 0.73 | 7 | 0.413 | 10 | 8.13 | 9 |
| TIP41 | 0.633 | 10 | 0.648 | 10 | 0.78 | 10 | 0.421 | 11 | 10.00 | 10 |
| TUA | 0.702 | 11 | 0.930 | 11 | 1.01 | 11 | 0.856 | 2 | 11.00 | 11 |
| Reference Gene | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EF1α | TUA | PP2A | F-BOX | UBI-1 | L23 | CYP | GAPDH | UBI-ep | CACS | TIP41 | |
| Ranking | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| n | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 16 |
| Geo Mean (Ct) | 20.21 | 22.19 | 24.13 | 26.67 | 24.24 | 21.48 | 18.62 | 18.27 | 20.04 | 24.99 | 26.10 |
| AR Mean (Ct) | 20.22 | 22.22 | 24.13 | 26.68 | 24.25 | 21.49 | 18.62 | 18.27 | 20.05 | 24.99 | 26.11 |
| Min (Ct) | 19.43 | 20.80 | 23.11 | 25.74 | 23.22 | 20.11 | 17.82 | 17.73 | 19.09 | 23.82 | 24.75 |
| Max (Ct) | 21.24 | 25.15 | 24.97 | 27.55 | 25.31 | 23.00 | 19.44 | 19.49 | 21.25 | 26.12 | 26.89 |
| SD (±Ct) | 0.40 | 0.81 | 0.48 | 0.41 | 0.53 | 0.47 | 0.31 | 0.36 | 0.41 | 0.43 | 0.54 |
| CV (%Ct) | 1.99 | 3.66 | 1.97 | 1.54 | 2.17 | 2.18 | 1.65 | 1.98 | 2.07 | 1.71 | 2.09 |
| Min (x-fold) | −1.72 | −2.63 | −2.02 | −1.91 | −2.03 | −2.59 | −1.74 | −1.45 | −1.94 | −2.24 | −2.55 |
| Max (x-fold) | 2.04 | 7.79 | 1.79 | 1.83 | 2.09 | 2.87 | 1.76 | 2.33 | 2.31 | 2.19 | 1.73 |
| SD (±x-fold) | 1.32 | 1.76 | 1.39 | 1.33 | 1.44 | 1.38 | 1.24 | 1.29 | 1.33 | 1.35 | 1.46 |
| coeff. of corr. (r) | 0.873 | 0.856 | 0.845 | 0.826 | 0.765 | 0.761 | 0.327 | 0.387 | 0.356 | 0.413 | 0.421 |
| p-value | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.216 | 0.138 | 0.176 | 0.112 | 0.105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Z.; Zhan, B.; Li, S. Selection and Validation of Reference Genes for Gene Expression Studies Using Quantitative Real-Time PCR in Prunus Necrotic Ringspot Virus-Infected Cucumis sativus. Viruses 2022, 14, 1269. https://doi.org/10.3390/v14061269
Dong Z, Zhan B, Li S. Selection and Validation of Reference Genes for Gene Expression Studies Using Quantitative Real-Time PCR in Prunus Necrotic Ringspot Virus-Infected Cucumis sativus. Viruses. 2022; 14(6):1269. https://doi.org/10.3390/v14061269
Chicago/Turabian StyleDong, Zhenfei, Binhui Zhan, and Shifang Li. 2022. "Selection and Validation of Reference Genes for Gene Expression Studies Using Quantitative Real-Time PCR in Prunus Necrotic Ringspot Virus-Infected Cucumis sativus" Viruses 14, no. 6: 1269. https://doi.org/10.3390/v14061269
APA StyleDong, Z., Zhan, B., & Li, S. (2022). Selection and Validation of Reference Genes for Gene Expression Studies Using Quantitative Real-Time PCR in Prunus Necrotic Ringspot Virus-Infected Cucumis sativus. Viruses, 14(6), 1269. https://doi.org/10.3390/v14061269