
The M13 Phage Assembly Machine Has a Membrane-Spanning Oligomeric Ring Structure


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phage and Plasmids
2.2. Purification of G1p/G11p
2.3. Circular Dichroism
2.4. Reconstitution into Liposomes
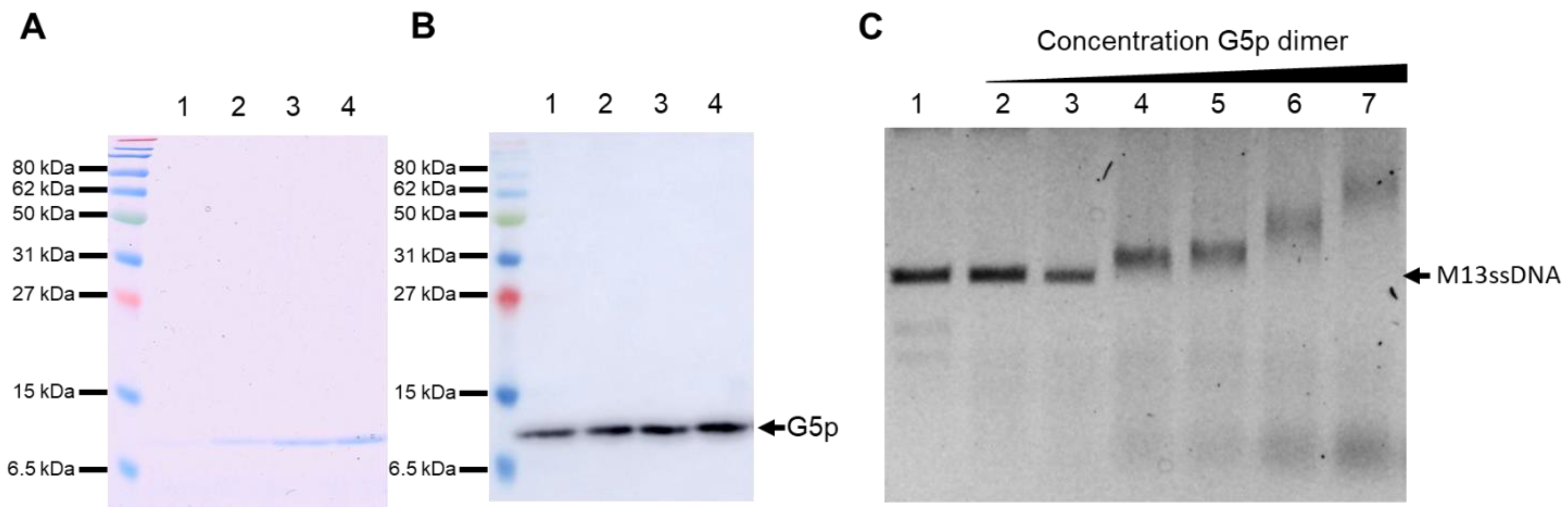
2.5. Purification of G5p-ssDNA
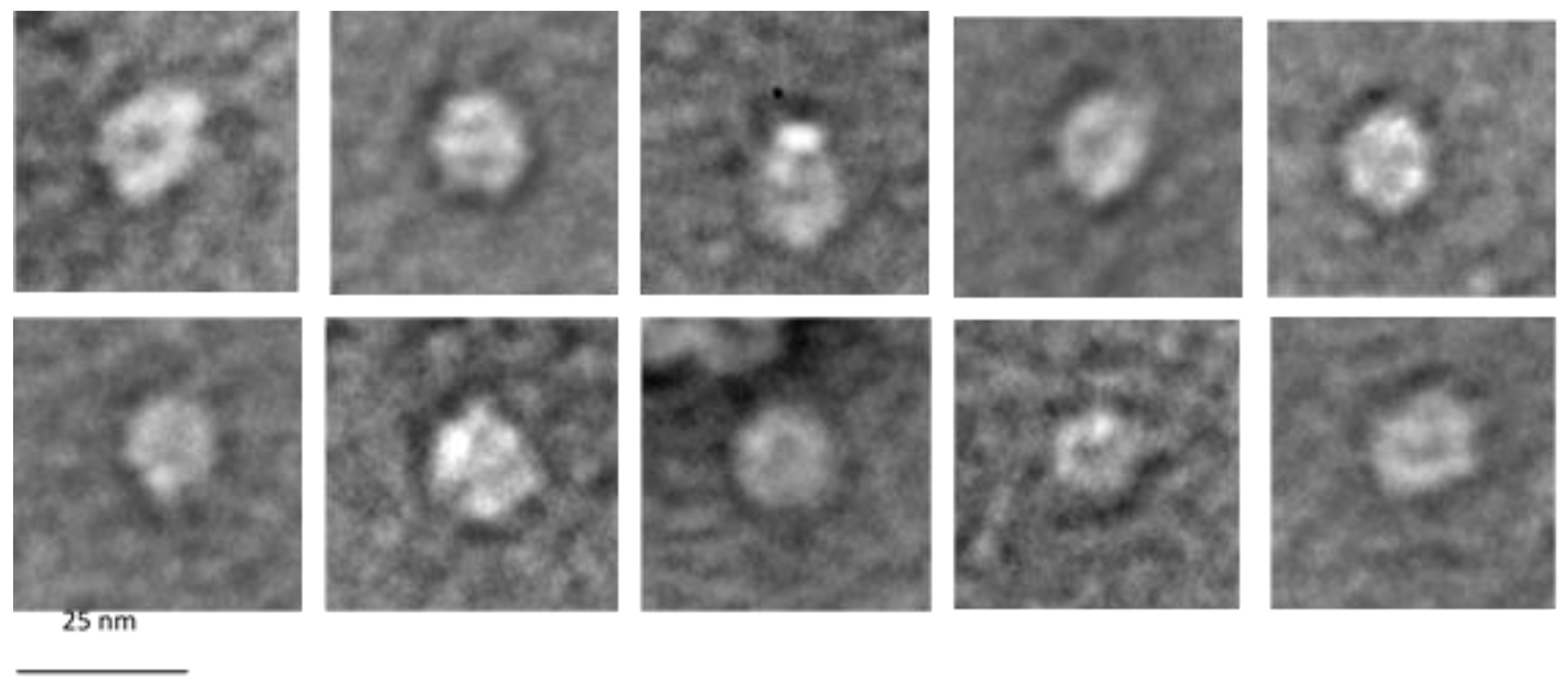
2.6. AFM and Electron Microscopy
3. Results
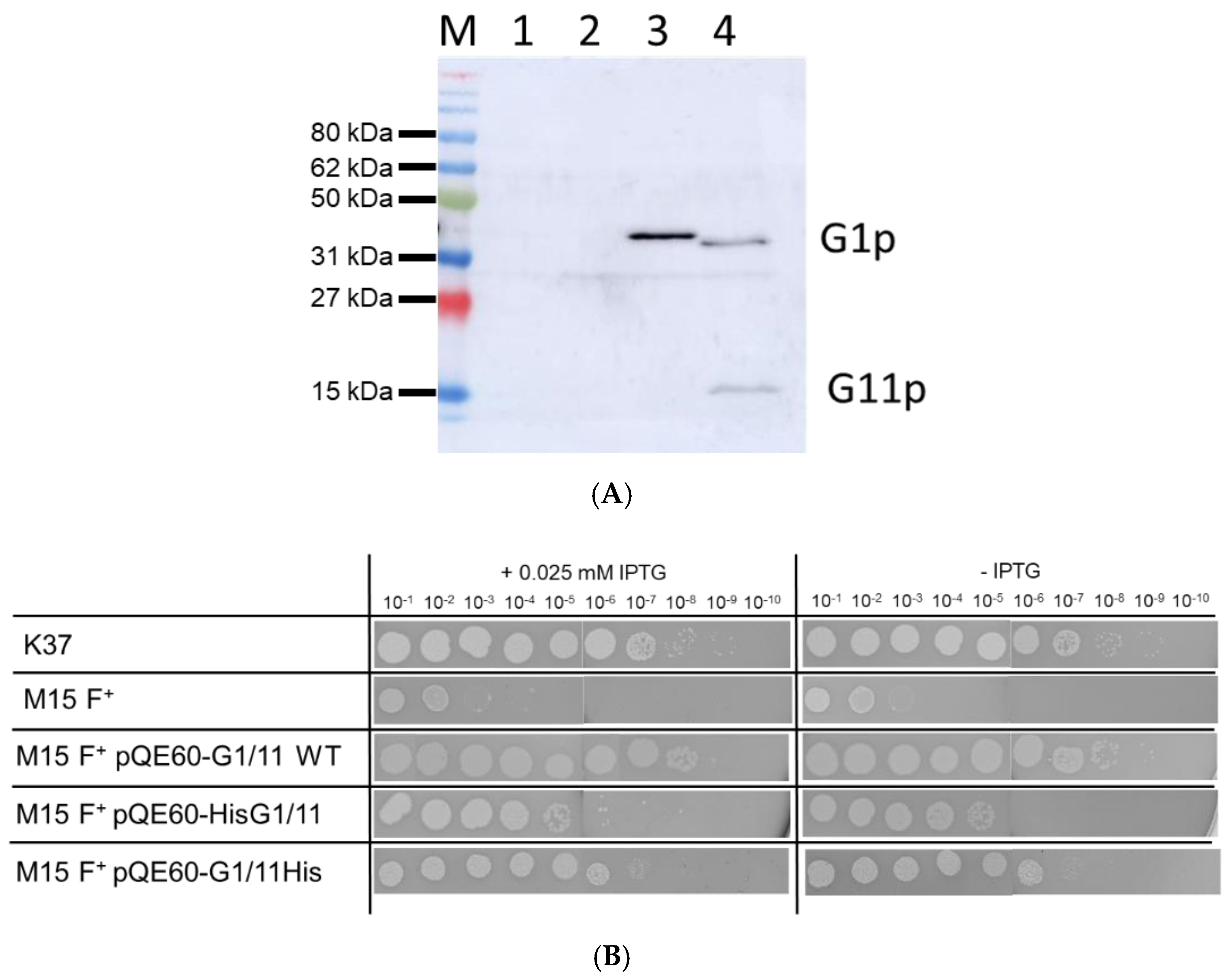
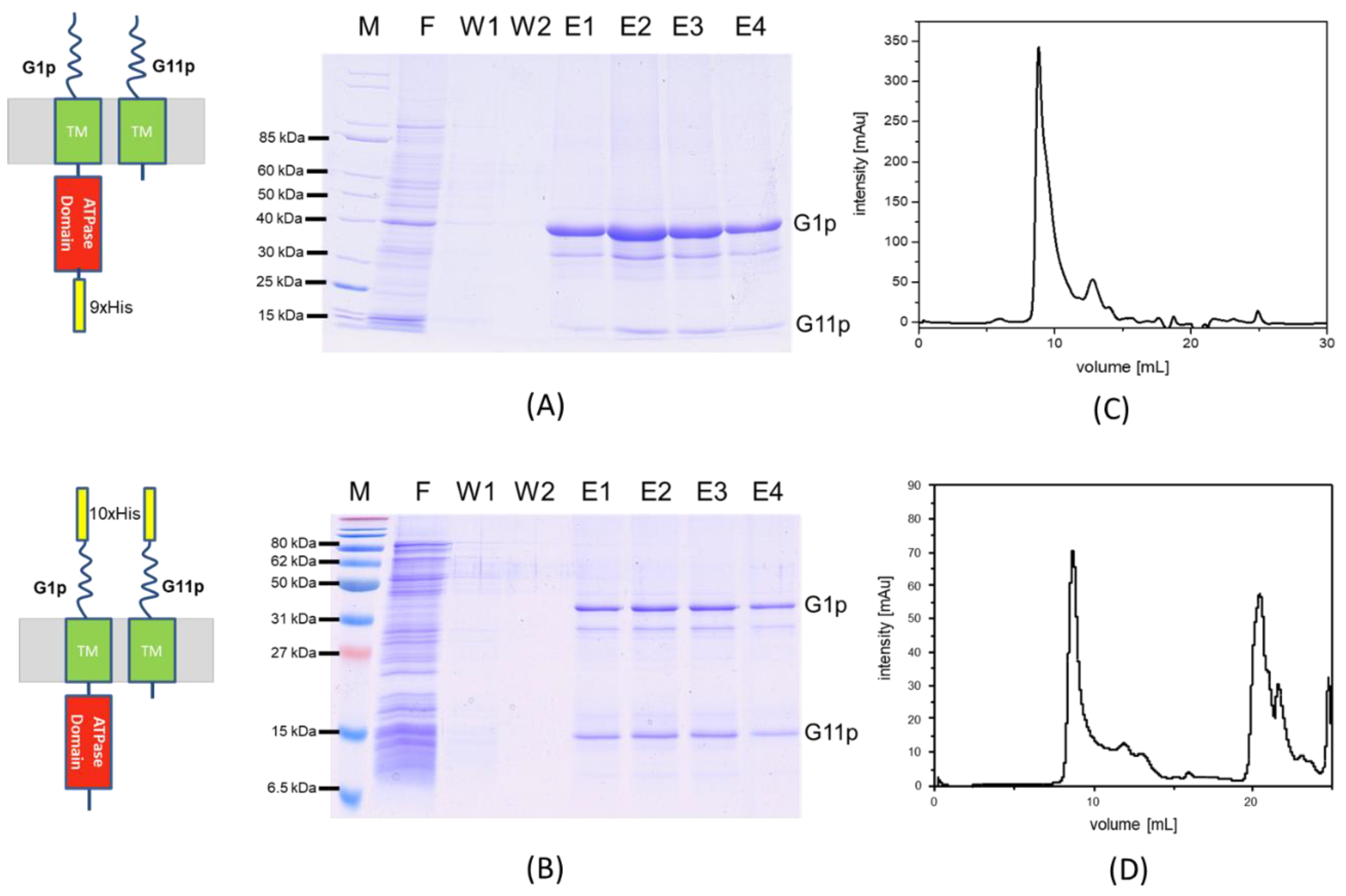
3.1. Expression of G1p and G11p
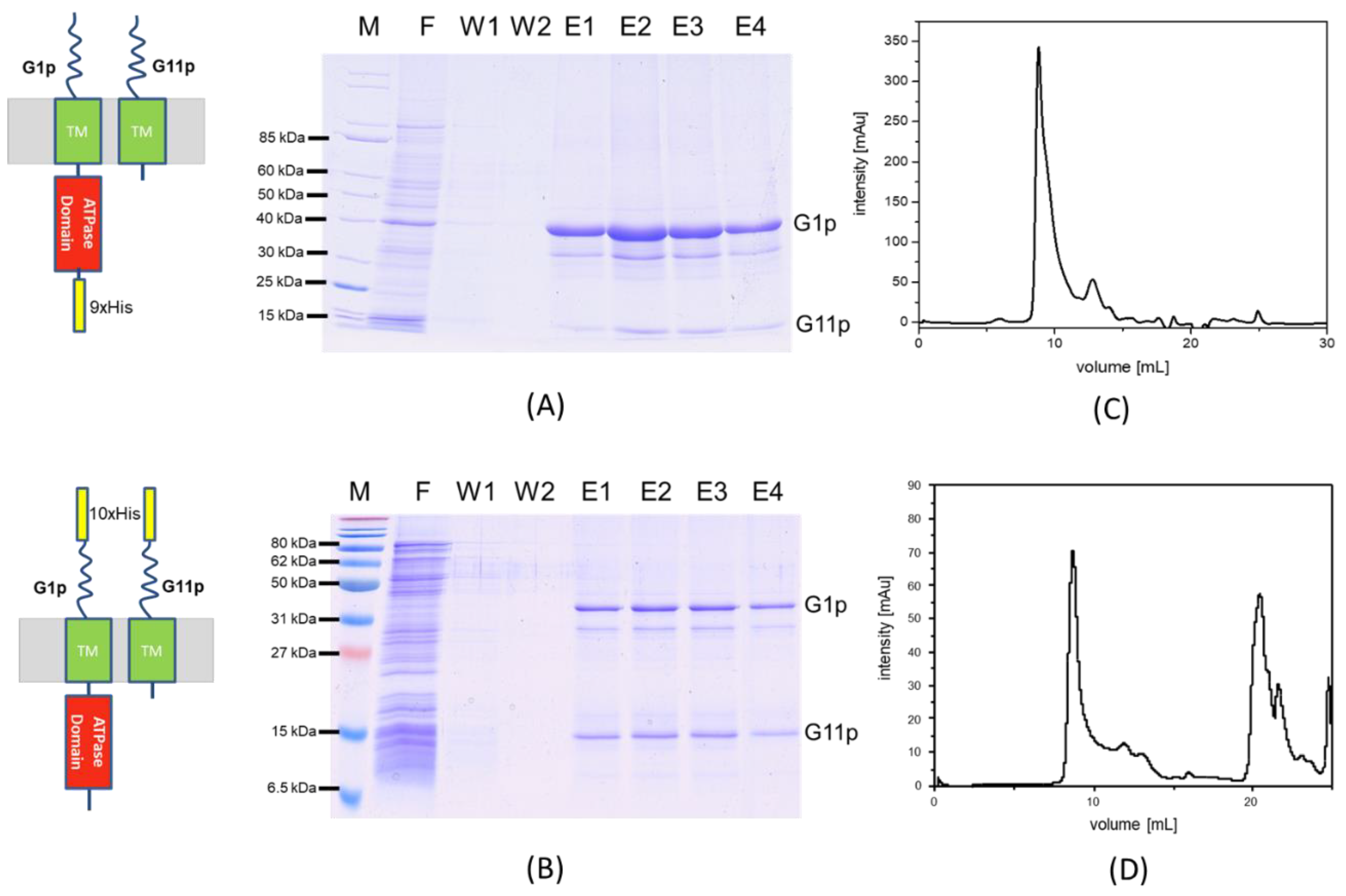
3.2. Purification of the M13 Assembly Machine
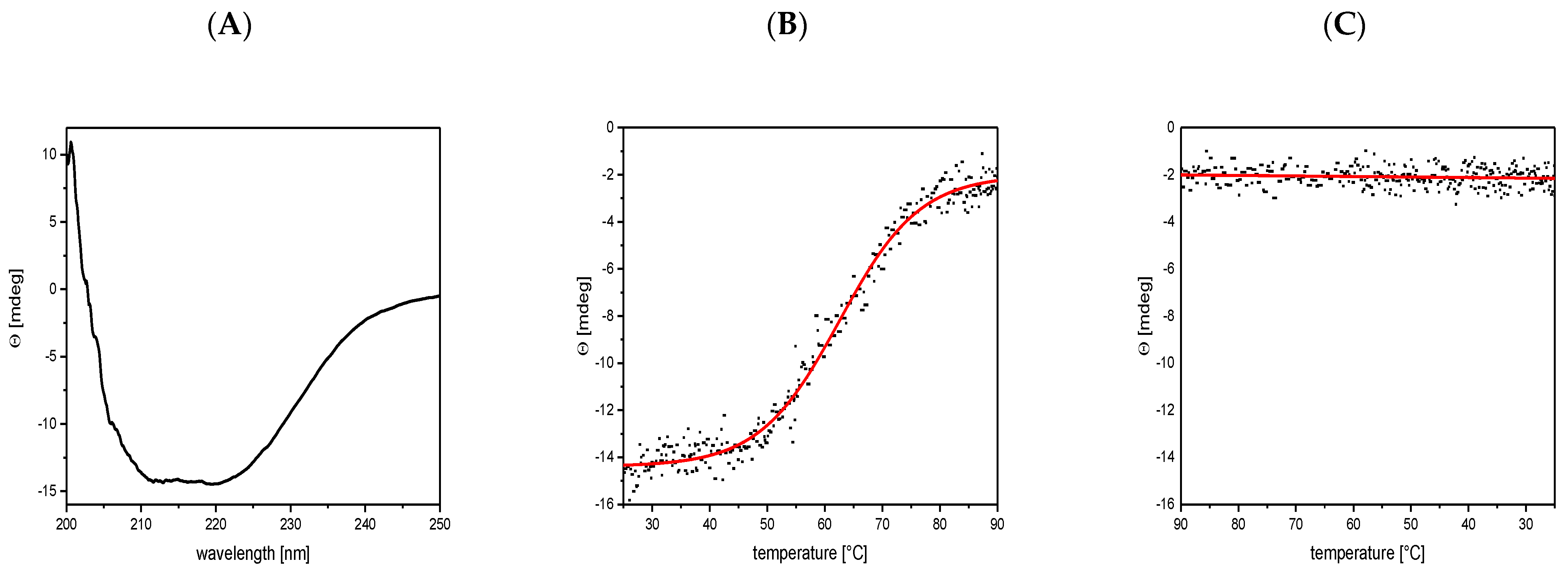
3.3. Circular Dichroism Shows a Mainly α-Helical Structure
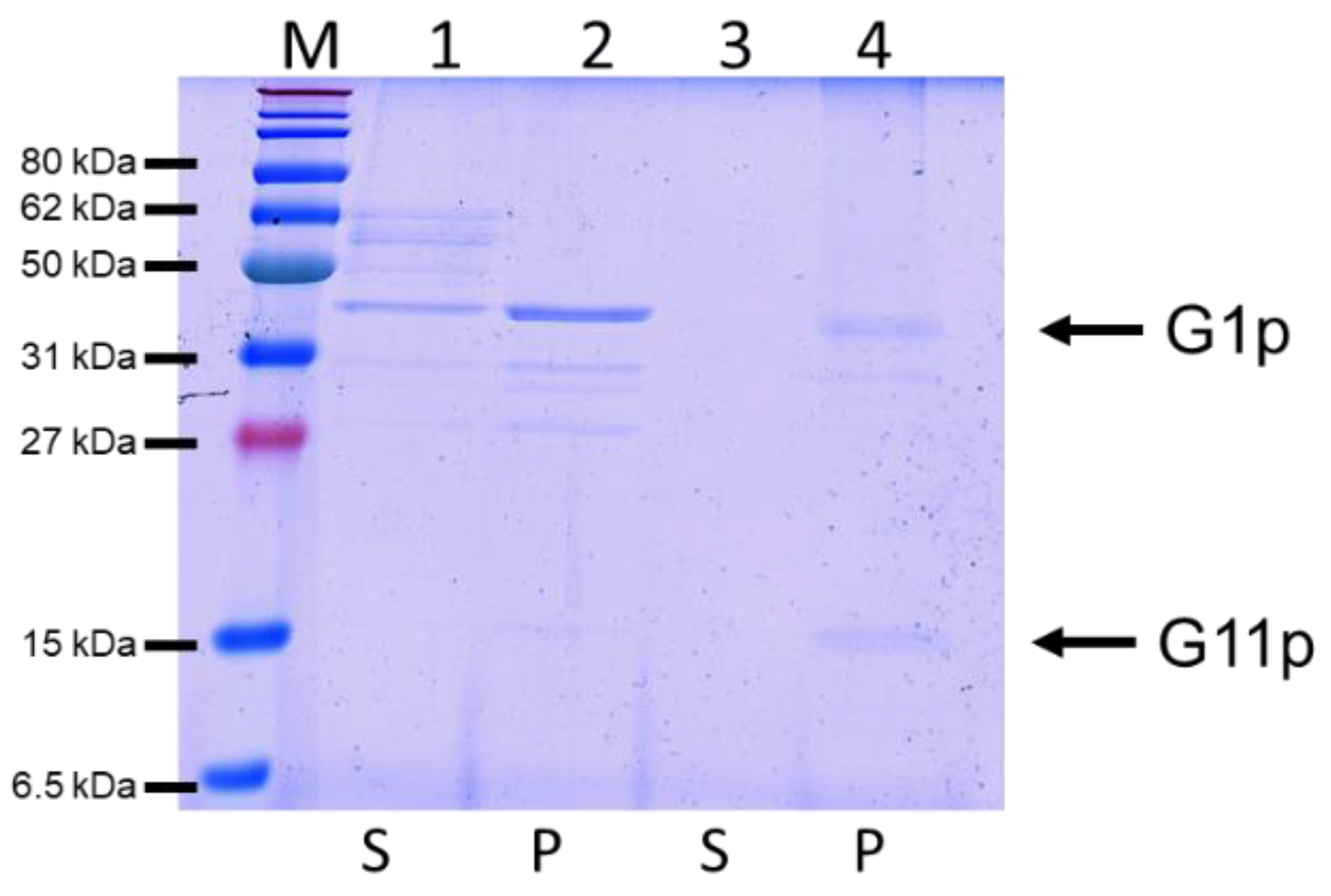
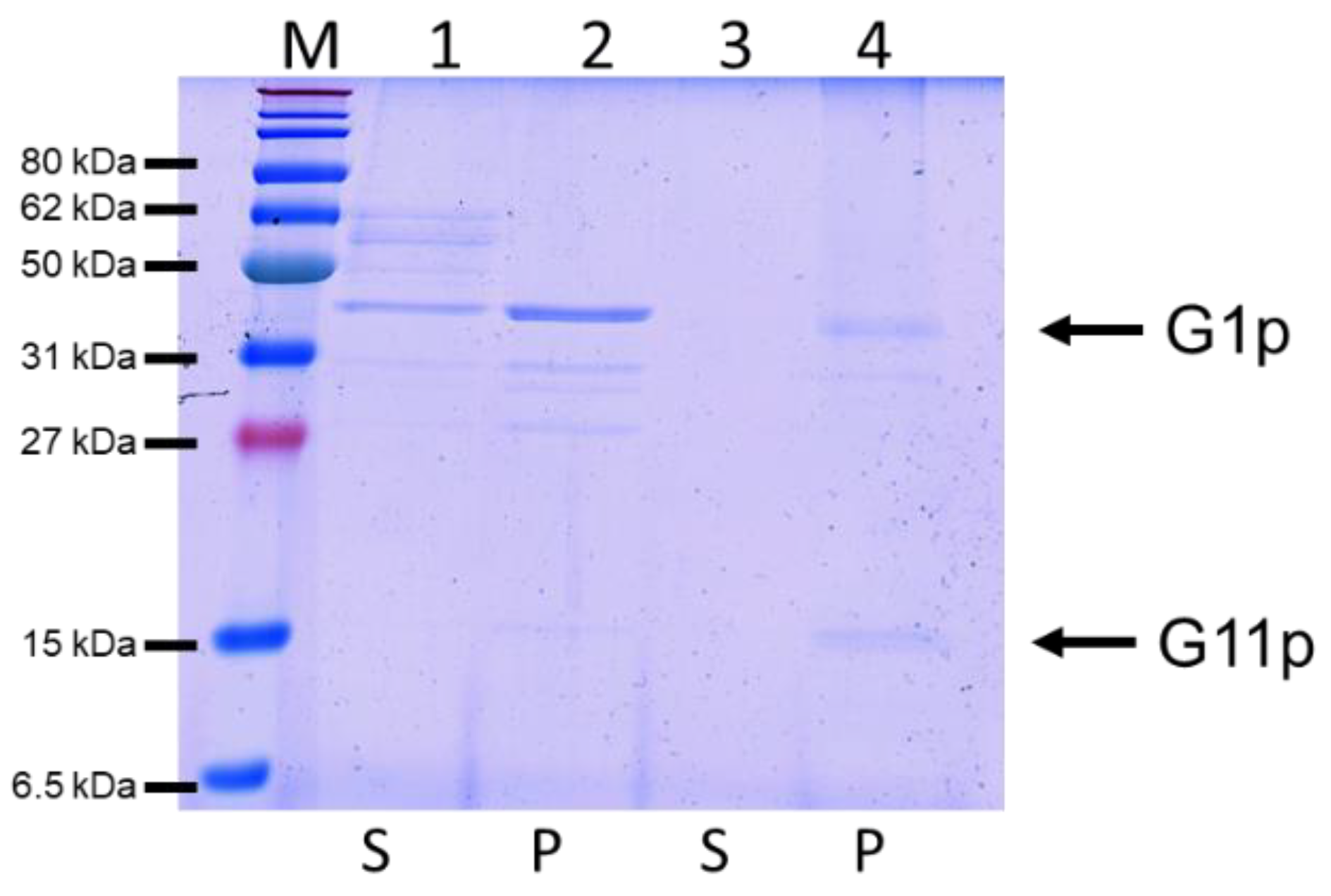
3.4. Reconstitution of G1p/G11p into Proteoliposomes
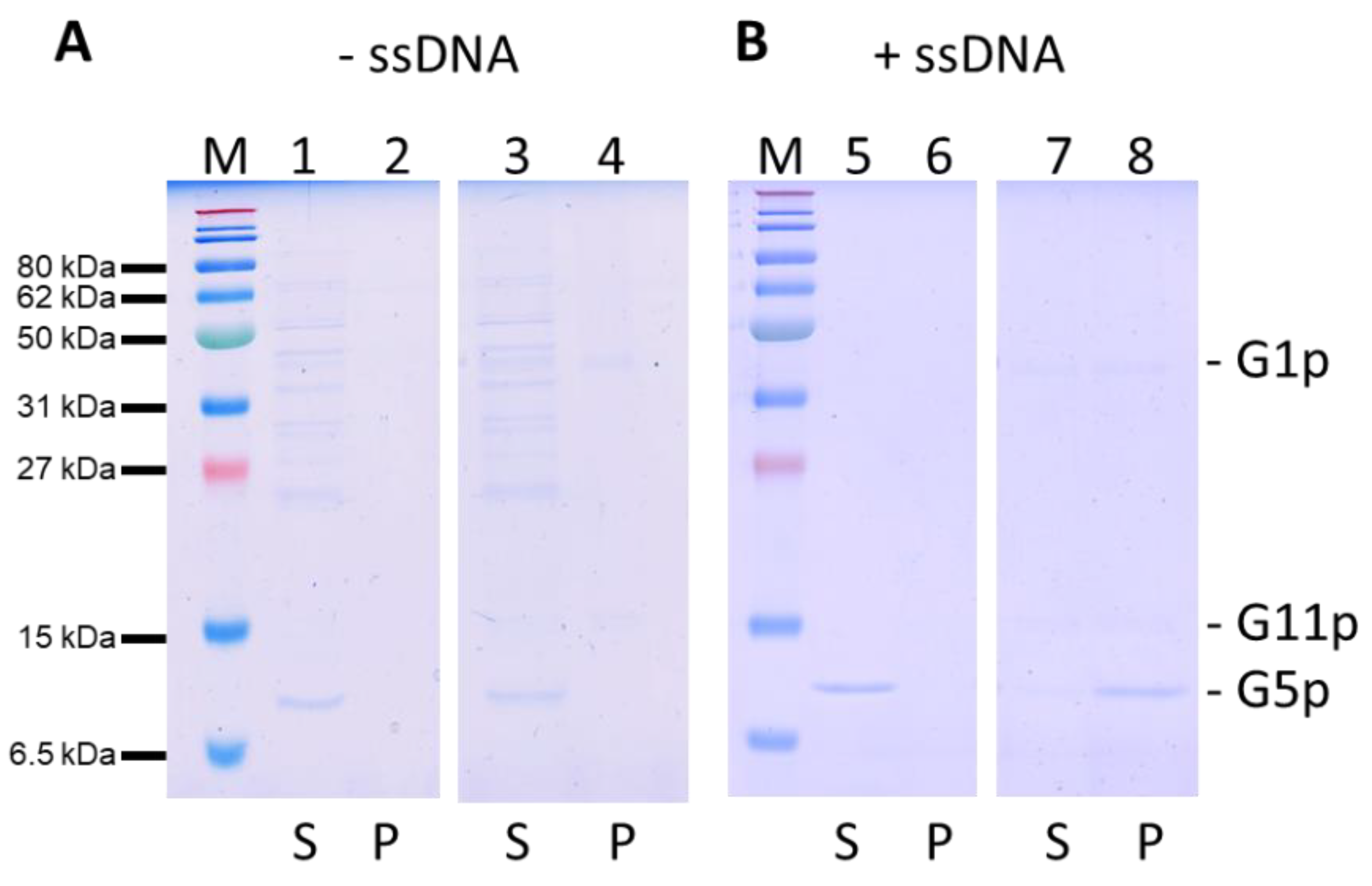
3.5. Purification and DNA Binding of the M13 G5p
3.6. Binding of the ssDNA to the G1p/G11p Complex
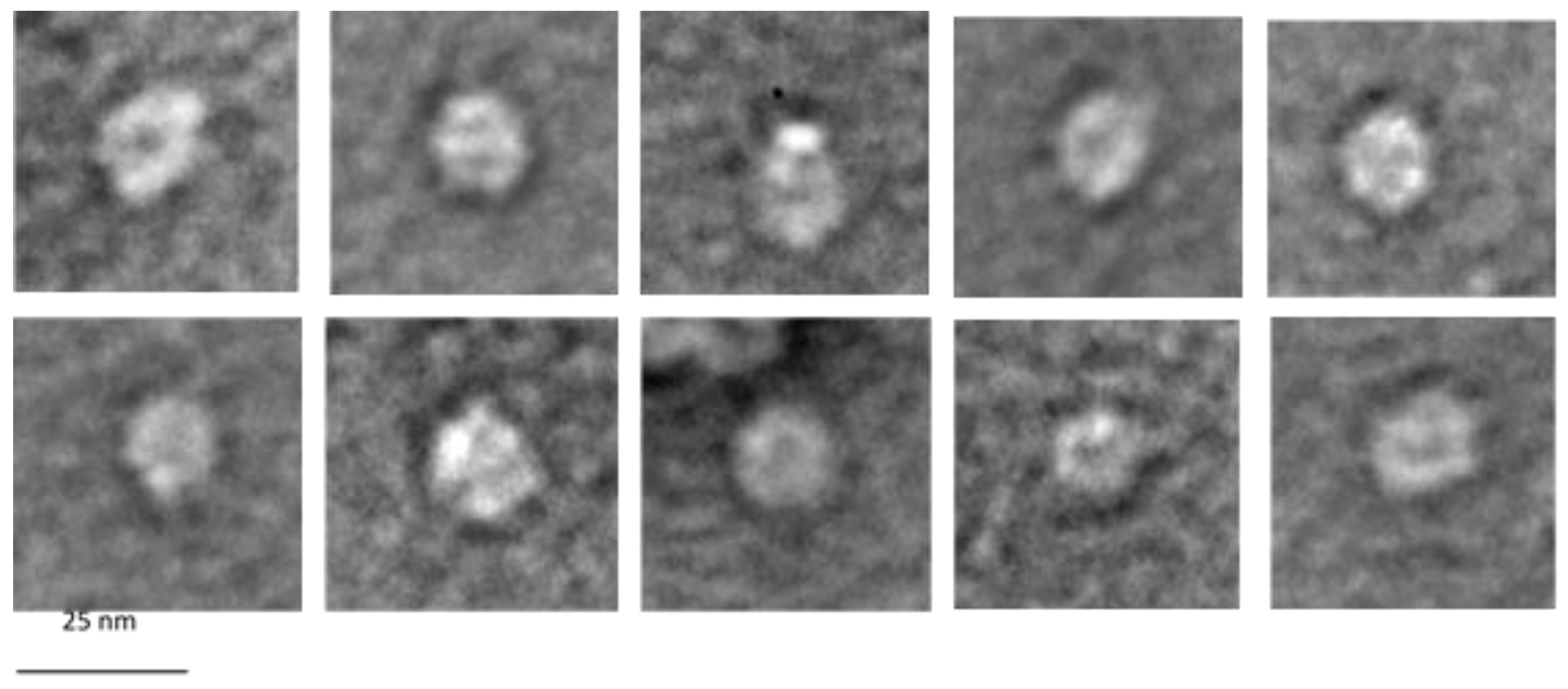
3.7. Electron Microscopy of G1p/G11p Complexes
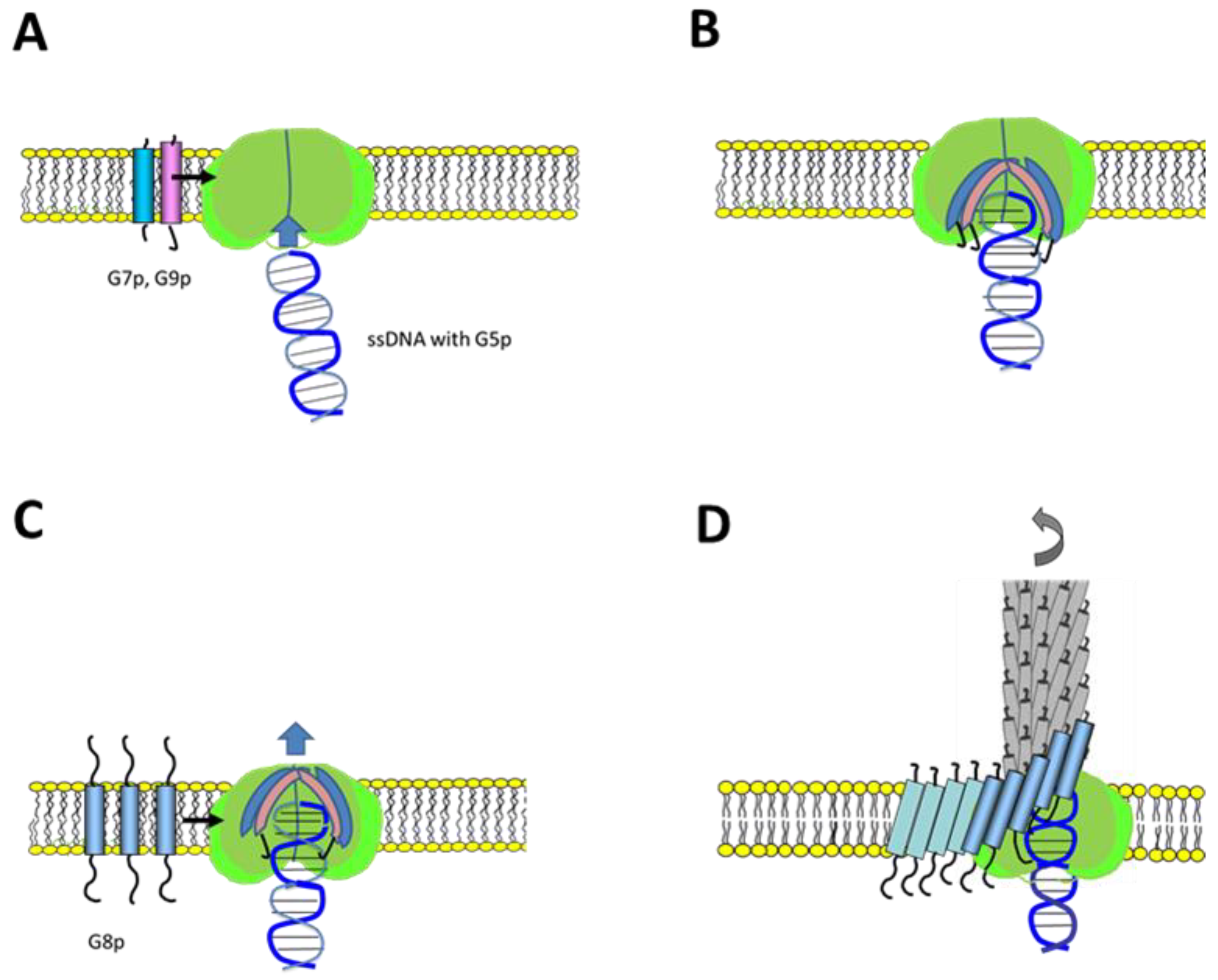
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuhn, A.; Leptihn, S. Helical and Filamentous Phages. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 53–60. ISBN 9780128145166. [Google Scholar]
- Ploss, M.; Kuhn, A. Kinetics of filamentous phage assembly. Phys. Biol. 2010, 7, 45002. [Google Scholar] [CrossRef] [PubMed]
- Loh, B.; Haase, M.; Mueller, L.; Kuhn, A.; Leptihn, S. The Transmembrane Morphogenesis Protein gp1 of Filamentous Phages Contains Walker A and Walker B Motifs Essential for Phage Assembly. Viruses 2017, 9, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoza, M.P.; Webster, R.E. The products of gene I and the overlapping in-frame gene XI are required for filamentous phage assembly. J. Mol. Biol. 1995, 248, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Ikoku, A.S.; Hearst, J.E. Identification of a structural hairpin in the filamentous chimeric phage M13Gori1. J. Mol. Biol. 1981, 151, 245–259. [Google Scholar] [CrossRef]
- Nagler, C.; Nagler, G.; Kuhn, A. Cysteine residues in the transmembrane regions of M13 procoat protein suggest that oligomeric coat proteins assemble onto phage progeny. J. Bacteriol. 2007, 189, 2897–2905. [Google Scholar] [CrossRef] [Green Version]
- Conners, R.; McLaren, M.; Łapińska, U.; Sanders, K.; Stone, M.R.L.; Blaskovich, M.A.T.; Pagliara, S.; Daum, B.; Rakonjac, J.; Gold, V.A.M. CryoEM structure of the outer membrane secretin channel pIV from the f1 filamentous bacteriophage. Nat. Commun. 2021, 12, 6316. [Google Scholar] [CrossRef]
- Kazmierczak, B.I.; Mielke, D.L.; Russel, M.; Model, P. pIV, a filamentous phage protein that mediates phage export across the bacterial cell envelope, forms a multimer. J. Mol. Biol. 1994, 238, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Perez-Iratxeta, C.; Andrade-Navarro, M.A. K2D2: Estimation of protein secondary structure from circular dichroism spectra. BMC Struct. Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Webster, R.E. Morphogenesis of filamentous bacteriophage f1: Orientation of extrusion and production of polyphage. Virology 1983, 127, 177–193. [Google Scholar] [CrossRef]
- Russel, M. Protein-protein interactions during filamentous phage assembly. J. Mol. Biol. 1993, 231, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.N.; Model, P.; Russel, M. A trans-envelope protein complex needed for filamentous phage assembly and export. Mol. Microbiol. 1999, 34, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Singh, D.; Lu, S.; Kottadiel, V.I.; Vafabakhsh, R.; Mahalingam, M.; Chemla, Y.R.; Ha, T.; Rao, V.B. A viral genome packaging ring-ATPase is a flexibly coordinated pentamer. Nat. Commun. 2021, 12, 6548. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Tang, W.-C.; Tao, P.; Mahalingam, M.; Fokine, A.; Rossmann, M.G.; Rao, V.B. Structural morphing in a symmetry-mismatched viral vertex. Nat. Commun. 2020, 11, 1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, D.; Laws, P.; Griffith, J. Complex of bacteriophage M13 single-stranded DNA and gene 5 protein. J. Mol. Biol. 1974, 82, 425–439. [Google Scholar] [CrossRef]
- Grant, R.A.; Webster, R.E. Minor protein content of the gene V protein/phage single-stranded DNA complex of the filamentous bacteriophage f1. Virology 1984, 133, 315–328. [Google Scholar] [CrossRef]
- Dotto, G.P.; Enea, V.; Zinderi, N.D. Functional analysis of bacteriophage f1 intergenic region. Virology 1981, 114, 463–473. [Google Scholar] [CrossRef]
- Houbiers, M.C.; Spruijt, R.B.; Demel, R.A.; Hemminga, M.A.; Wolfs, C.J.A.M. Spontaneous insertion of gene 9 minor coat protein of bacteriophage M13 in model membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2001, 1511, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Ploss, M.; Kuhn, A. Membrane insertion and assembly of epitope-tagged gp9 at the tip of the M13 phage. BMC Microbiol. 2011, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Løset, G.Å.; Roos, N.; Bogen, B.; Sandlie, I. Expanding the versatility of phage display II: Improved affinity selection of folded domains on protein VII and IX of the filamentous phage. PLoS ONE 2011, 6, e17433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, G.J.; Rowitch, D.H.; Perham, R.N. Interactions between DNA and coat protein in the structure and assembly of filamentous bacteriophage fd. Nature 1987, 327, 252–254. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haase, M.; Tessmer, L.; Köhnlechner, L.; Kuhn, A. The M13 Phage Assembly Machine Has a Membrane-Spanning Oligomeric Ring Structure. Viruses 2022, 14, 1163. https://doi.org/10.3390/v14061163
Haase M, Tessmer L, Köhnlechner L, Kuhn A. The M13 Phage Assembly Machine Has a Membrane-Spanning Oligomeric Ring Structure. Viruses. 2022; 14(6):1163. https://doi.org/10.3390/v14061163
Chicago/Turabian StyleHaase, Maximilian, Lutz Tessmer, Lilian Köhnlechner, and Andreas Kuhn. 2022. "The M13 Phage Assembly Machine Has a Membrane-Spanning Oligomeric Ring Structure" Viruses 14, no. 6: 1163. https://doi.org/10.3390/v14061163
APA StyleHaase, M., Tessmer, L., Köhnlechner, L., & Kuhn, A. (2022). The M13 Phage Assembly Machine Has a Membrane-Spanning Oligomeric Ring Structure. Viruses, 14(6), 1163. https://doi.org/10.3390/v14061163