
Memory CD8 T Cells Protect against Cytomegalovirus Disease by Formation of Nodular Inflammatory Foci Preventing Intra-Tissue Virus Spread

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice, Virus, and the Route of Infection
2.2. Adoptive Transfer of Memory CD8 T Cells (CD8-AT)
2.3. Peptides and Quantitation of Functional Epitope-Specific Memory CD8 T Cells
2.4. Cytofluorometric Analyses
2.5. Quantitation of Tissue Infection and CD8 T Cell Infiltration
2.6. In Situ Detection of Proliferating CD8 T Cells
2.7. Determination of Viral and Cellular Doubling Times and Number of Cell Divisions
3. Results and Discussion
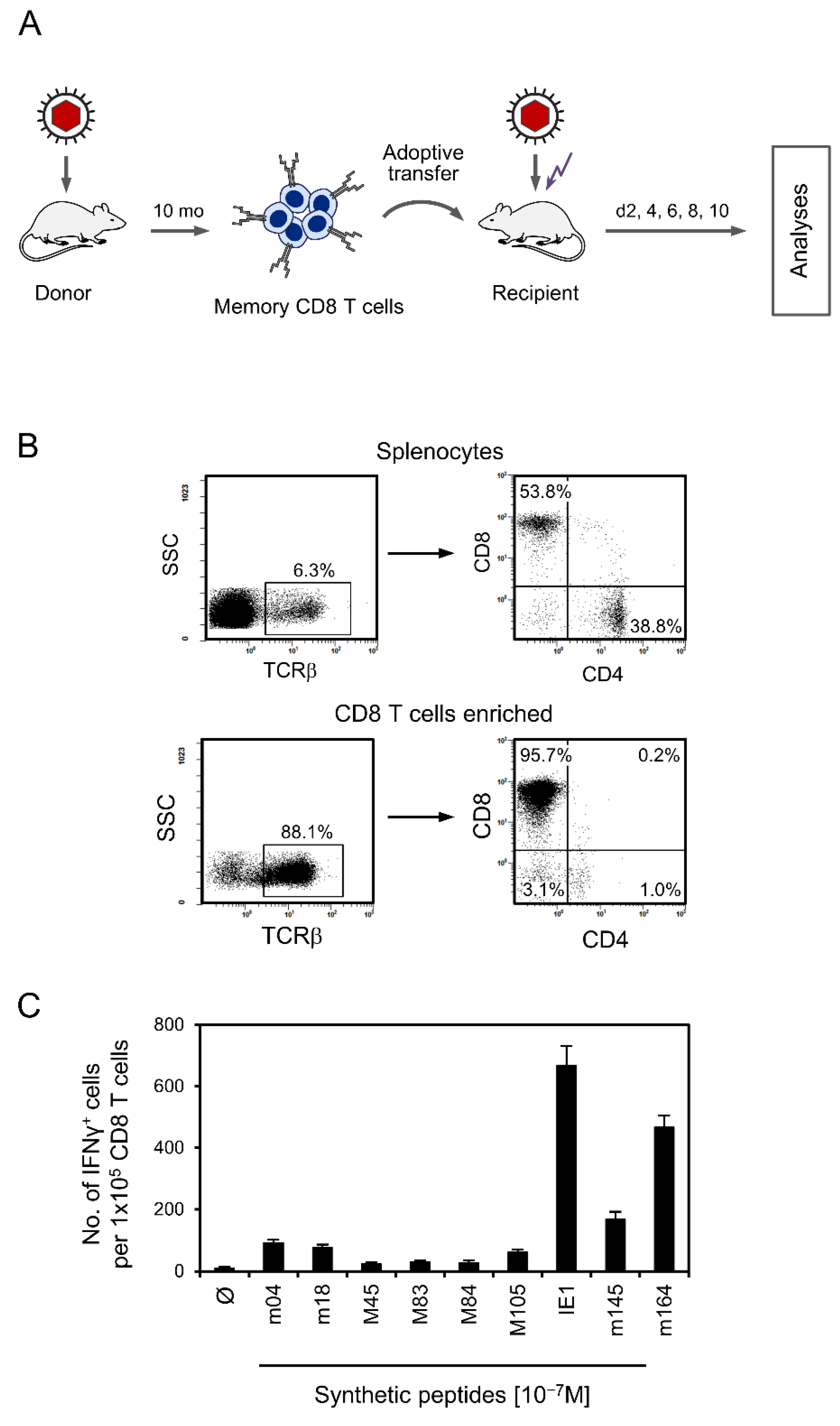
3.1. Experimental Protocol of CD8-AT and Specificity Composition of the Transferred Cells
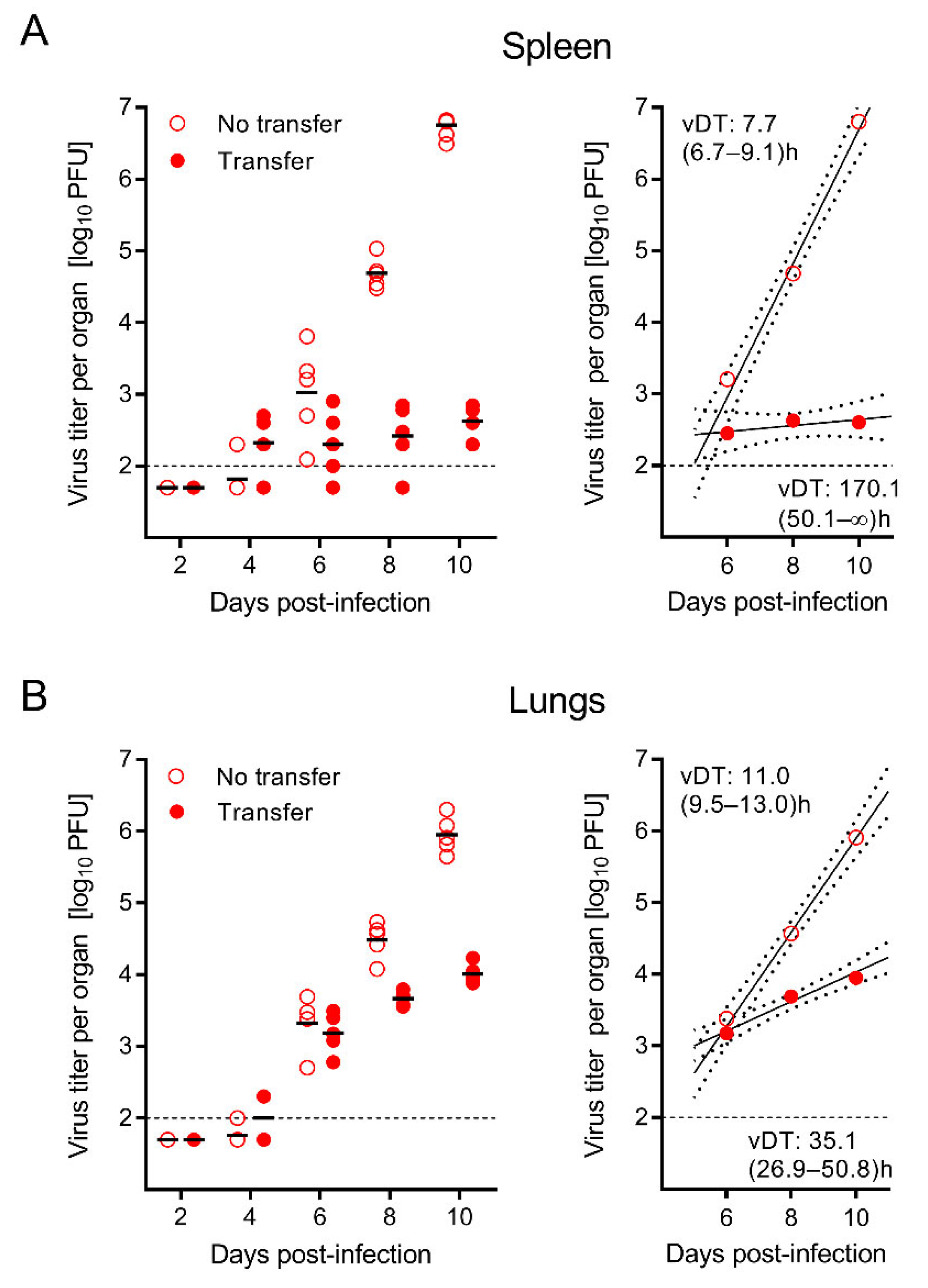
3.2. The Time Course of Infection after CD8-AT Reveals Inhibition of Intra-Tissue Viral Spread
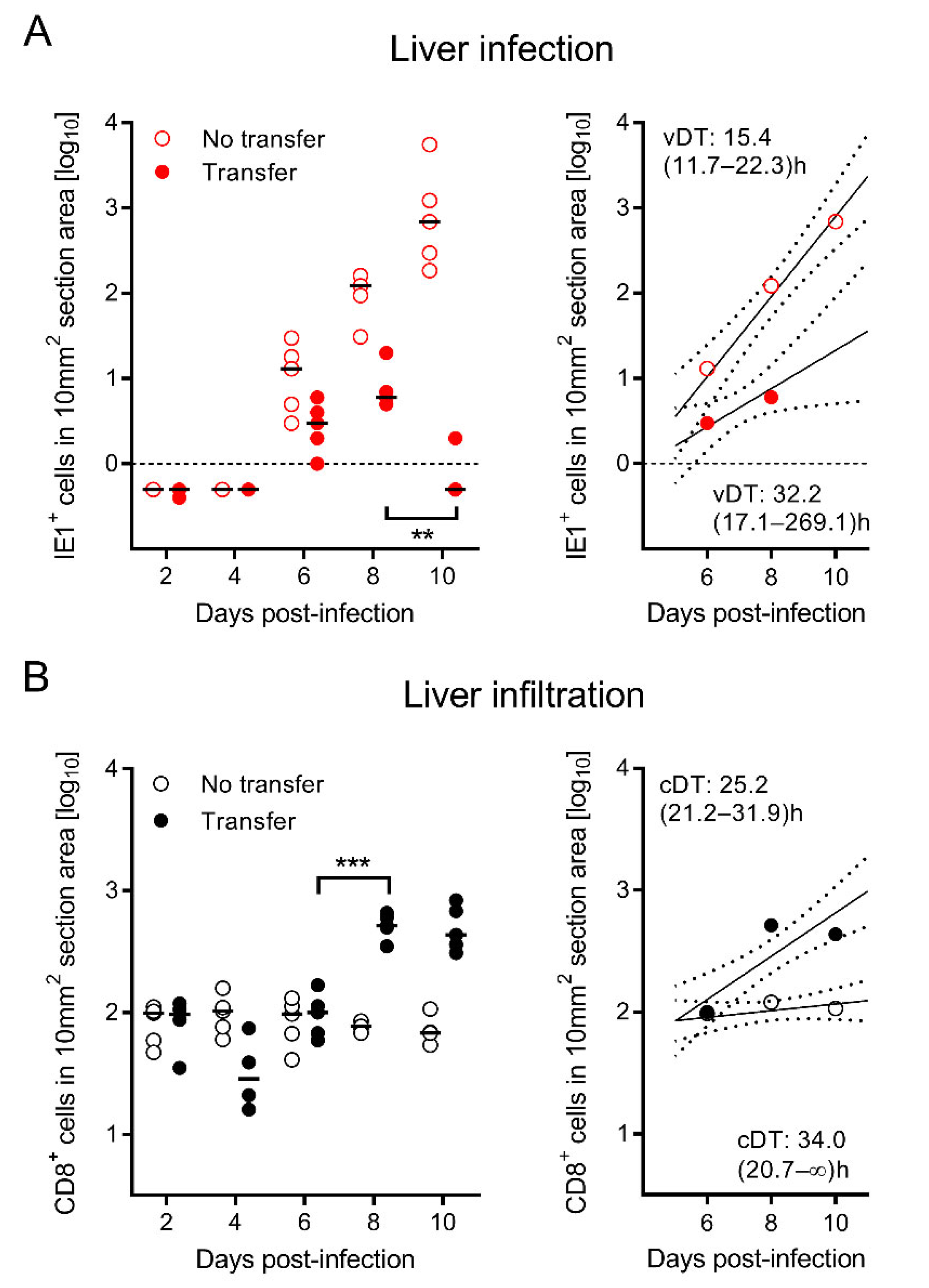
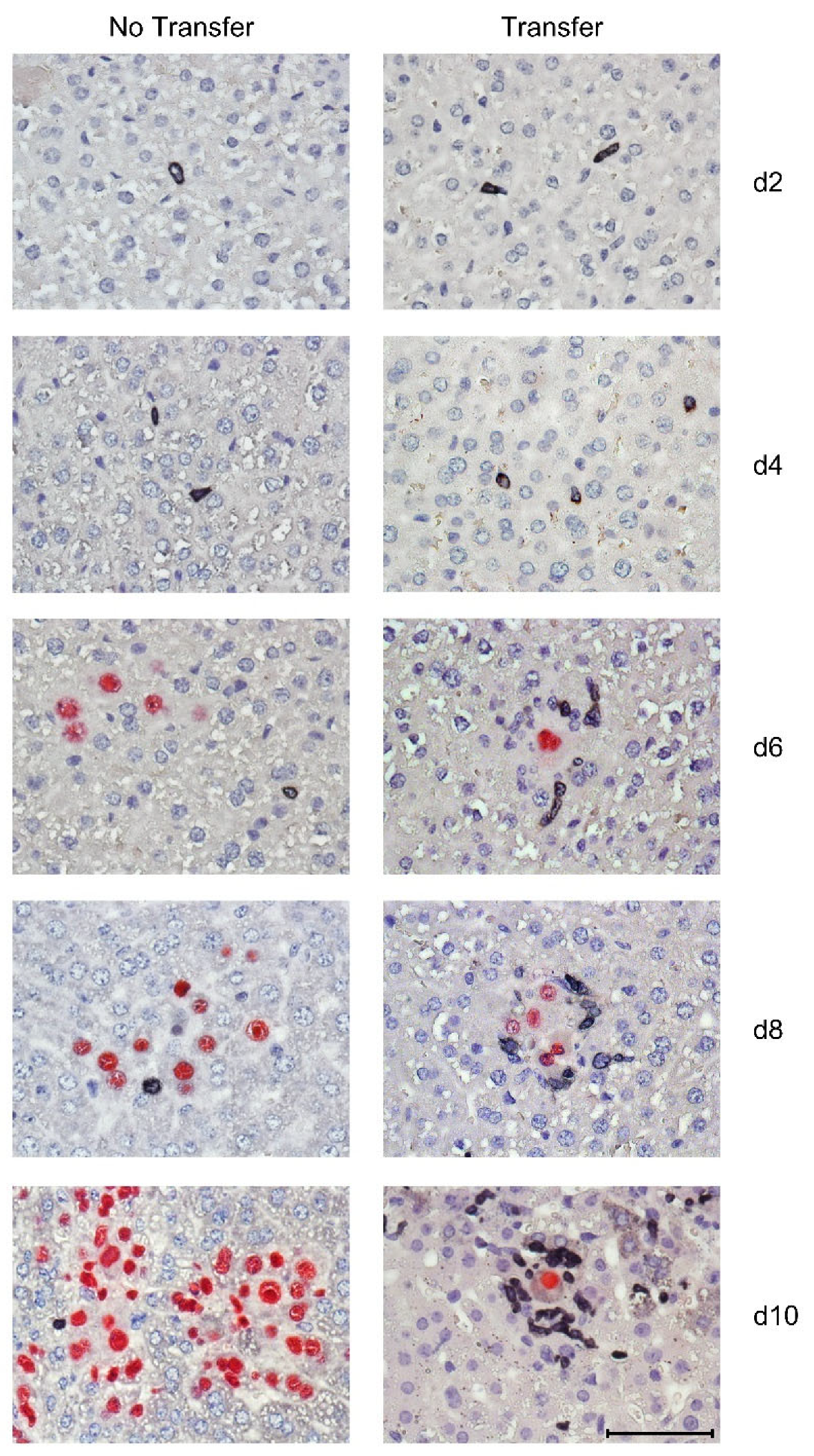
3.3. Tissue-Infiltrating CD8 T Cells Confine Infection to Nodular Inflammatory Foci (NIF)
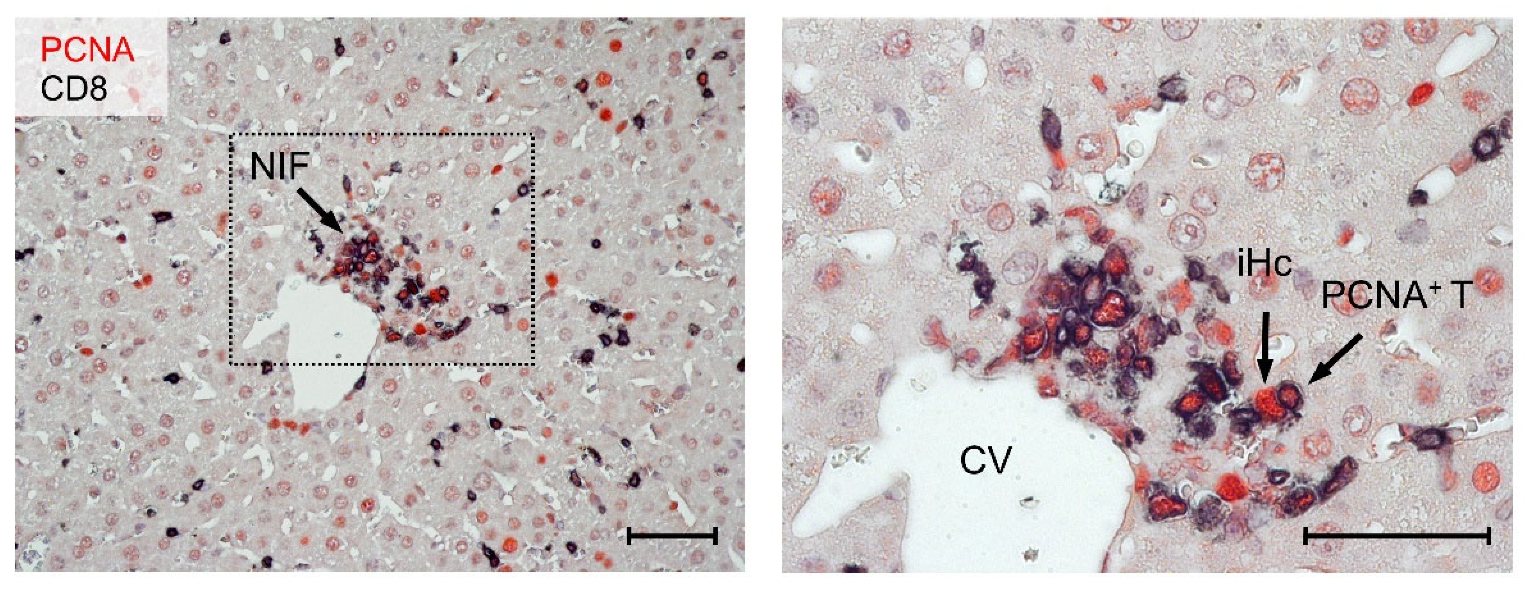
3.4. Most CD8 T Cells within NIF Express Proliferating Cell nuclear Antigen (PCNA)
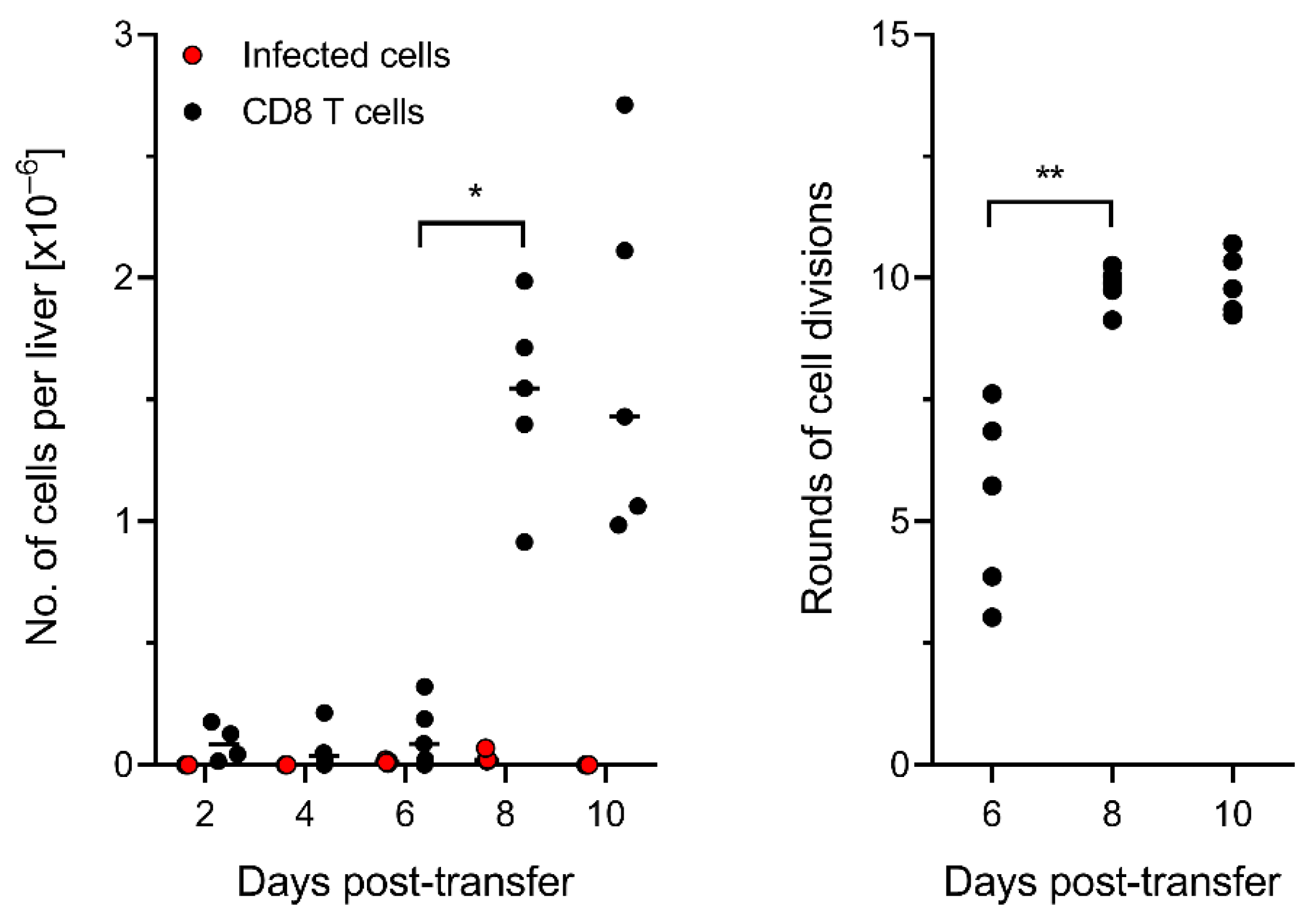
3.5. Control of Intra-Tissue Virus Spread Corresponds to Extensive CD8 T-Cell Expansion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 1–22. [Google Scholar]
- Smith, M.G. Propagation of salivary gland virus of the mouse in tissue cultures. Proc. Soc. Exp. Biol. Med. 1954, 86, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.G. Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc. Soc. Exp. Biol. Med. 1956, 92, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Pawletko, K.; Indenbirken, D.; Schumacher, U.; Brune, W. Stepwise adaptation of murine cytomegalovirus to cells of a foreign host for identification of host range determinants. Med. Microbiol. Immunol. 2015, 204, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Kaneshima, H.; Mocarski, E.S. Dramatic interstrain differences in the replication of human cytomegalovirus in SCID-hu mice. J. Infect. Dis. 1995, 171, 1599–1603. [Google Scholar] [CrossRef]
- Smith, M.S.; Streblow, D.N.; Caposio, P.; Nelson, J.A. Humanized mouse models of cytomegalovirus pathogenesis and latency. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 417–436. [Google Scholar]
- Crawford, L.B.; Streblow, D.N.; Hakki, M.; Nelson, J.A.; Caposio, P. Humanized mouse models of human cytomegalovirus infection. Curr. Opin. Virol. 2015, 13, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Lemmermann, N.A.W. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: Predictions for medical translation that survived the “test of time”. Viruses 2018, 10, e693. [Google Scholar] [CrossRef] [Green Version]
- Powers, C.; Früh, K. Rhesus CMV: An emerging animal model for human CMV. Med. Microbiol. Immunol. 2008, 197, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Wang, Z.; Abel, K.; Li, J.; Strelow, L.; Mandarino, A.; Eberhardt, M.K.; Schmidt, K.A.; Diamond, D.J.; Barry, P.A. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med. Microbiol. Immunol. 2008, 197, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Früh, K.; Malouli, D.; Oxford, K.L.; Barry, P.A. Non-human-primate models of cytomegalovirus infection, prevention, and therapy. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 461–494. [Google Scholar]
- Deere, J.D.; Barry, P.A. Using the nonhuman primate model of HCMV to guide vaccine development. Viruses 2014, 6, 1483–1501. [Google Scholar] [CrossRef] [Green Version]
- Itell, H.L.; Kaur, A.; Deere, J.D.; Barry, P.A.; Permar, S.R. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr. Opin. Virol. 2017, 25, 126–133. [Google Scholar] [CrossRef] [Green Version]
- McGregor, A.; McVoy, M.A.; Schleiss, M.R. The Guinea pig model of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 88–118. [Google Scholar]
- Slavuljica, I.; Kveštak, D.; Huszthy, P.C.; Kosmac, K.; Britt, W.J.; Jonjić, S. Immunobiology of congenital cytomegalovirus infection of the central nervous system—The murine cytomegalovirus model. Cell. Mol. Immunol. 2015, 12, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, S.; Ettinger, J.; Streblow, D.N. The rat model of cytomegalovirus infection and vascular disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 312–336. [Google Scholar]
- Benedict, C.A.; Crozat, K.; Degli-Esposti, M.; Dalod, M. Host genetic models in cytomegalovirus immunology. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 310–334. [Google Scholar]
- Ruzsics, Z.; Borst, E.M.; Bosse, J.B.; Brune, W.; Messerle, M. Manipulating CMV genomes by BAC mutagenesis: Strategies and applications. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 38–58. [Google Scholar]
- Lemmermann, N.A.; Kropp, K.A.; Seckert, C.K.; Grzimek, N.K.; Reddehase, M.J. Reverse genetics modification of cytomegalovirus antigenicity and immunogenicity by CD8 T-cell epitope deletion and insertion. J. Biomed. Biotechnol. 2011, 2011, 812742. [Google Scholar] [CrossRef] [PubMed]
- Redwood, A.J.; Shellam, G.R.; Smith, L.M. Molecular evolution of murine cytomegalovirus genomes. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 23–37. [Google Scholar]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobedman, B.; Mocarski, E.S. Quantitative analysis of latent human cytomegalovirus. J. Virol. 1999, 73, 4806–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.; Sinclair, J. Epigenetic regulation of human cytomegalovirus gene expression: Impact on latency and reactivation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 330–346. [Google Scholar]
- Poole, E.; Sinclair, J. Sleepless latency of human cytomegalovirus. Med. Microbiol. Immunol. 2015, 204, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, F. Human cytomegalovirus latency: Approaching the Gordian Knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [Green Version]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. mBio 2018, 9, e00013-18. [Google Scholar] [CrossRef] [Green Version]
- Elder, E.; Sinclair, J. HCMV latency: What regulates the regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Griessl, M.; Renzaho, A.; Freitag, K.; Seckert, C.K.; Reddehase, M.J.; Lemmermann, N.A.W. Stochastic episodes of latent cytomegalovirus transcription drive CD8 T-cell “memory inflation” and avoid immune evasion. Front. Immunol. 2021, 12, 668885. [Google Scholar] [CrossRef] [PubMed]
- Mayo, D.R.; Armstrong, J.A.; Ho, M. Reactivation of murine cytomegalovirus by cyclophosphamide. Nature 1977, 267, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Balthesen, M.; Messerle, M.; Reddehase, M.J. Lungs are a major organ site of cytomegalovirus latency and recurrence. J. Virol. 1993, 67, 5360–5366. [Google Scholar] [CrossRef] [Green Version]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.S.; Goldman, D.C.; Bailey, A.S.; Pfaffle, D.L.; Kreklywich, C.N.; Spencer, D.B.; Othieno, F.A.; Streblow, D.N.; Garcia, J.V.; Fleming, W.H.; et al. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 2010, 8, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakki, M.; Goldman, D.C.; Streblow, D.N.; Hamlin, K.L.; Krekylwich, C.N.; Fleming, W.H.; Nelson, J.A. HCMV infection of humanized mice after transplantation of G-CSF-mobilized peripheral blood stem cells from HCMV-seropositive donors. Biol. Blood Marrow. Transpl. 2014, 20, 132–135. [Google Scholar] [CrossRef] [Green Version]
- Plachter, B.; Sinzger, C.; Jahn, G. Cell types involved in replication and distribution of human cytomegalovirus. Adv. Virus Res. 1996, 46, 195–261. [Google Scholar] [CrossRef]
- Podlech, J.; Holtappels, R.; Wirtz, N.; Steffens, H.P.; Reddehase, M.J. Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J. Gen. Virol. 1998, 79, 2099–2104. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 2019, 208, 495–512. [Google Scholar] [CrossRef]
- Lisnić, B.; Lisnić, V.J.; Jonjić, S. NK cell interplay with cytomegaloviruses. Curr. Opin. Virol. 2015, 15, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat. Rev. Immunol. 2020, 20, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.; Hill, A.; Tortorella, D.; Ploegh, H. Cytomegaloviruses use multiple mechanisms to elude the host immune response. Immunol. Lett. 1997, 57, 213–216. [Google Scholar] [CrossRef]
- Hengel, H.; Reusch, U.; Gutermann, A.; Ziegler, H.; Jonjic, S.; Lucin, P.; Koszinowski, U.H. Cytomegaloviral control of MHC class I function in the mouse. Immunol. Rev. 1999, 168, 167–176. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Früh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar] [CrossRef]
- Doom, C.M.; Hill, A.B. MHC class I immune evasion in MCMV infection. Med. Microbiol. Immunol. 2008, 197, 191–204. [Google Scholar] [CrossRef]
- Becker, S.; Fink, A.; Podlech, J.; Reddehase, M.J.; Lemmermann, N.A. Host-adapted gene families involved in murine cytomegalovirus immune evasion. Viruses 2022, 14, 128. [Google Scholar] [CrossRef]
- Cannon, M.J.; Grosse, S.D.; Fowler, K.B. The epidemiology and public health impact of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 26–48. [Google Scholar]
- Adler, S.P.; Nigro, G. Clinical cytomegalovirus research: Congenital infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 55–73. [Google Scholar]
- Ho, M.; Suwansirikul, S.; Dowling, J.N.; Youngblood, L.A.; Armstrong, J.A. The transplanted kidney as a source of cytomegalovirus infection. N. Engl. J Med. 1975, 293, 1109–1112. [Google Scholar] [CrossRef]
- Chou, S.W. Acquisition of donor strains of cytomegalovirus by renal-transplant recipients. N. Engl. J. Med. 1986, 314, 1418–1423. [Google Scholar] [CrossRef]
- Grundy, J.E.; Lui, S.F.; Super, M.; Berry, N.J.; Sweny, P.; Fernando, O.N.; Moorhead, J.; Griffiths, P.D. Symptomatic cytomegalovirus infection in seropositive kidney recipients: Reinfection with donor virus rather than reactivation of recipient virus. Lancet 1988, 2, 132–135. [Google Scholar] [CrossRef]
- Emery, V.C. Relative importance of cytomegalovirus load as a risk factor for cytomegalovirus disease in the immunocompromised host. Monogr. Virol. 1998, 21, 288–301. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Lemmermann, N.A.W. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J. Mutual interference between cytomegalovirus and reconstitution of protective immunity after hematopoietic cell transplantation. Front. Immunol. 2016, 7, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddehase, M.J.; Holtappels, R.; Lemmermann, N.A.W. Consequence of histoincompatibility beyond GvH-reaction in cytomegalovirus disease associated with allogeneic hematopoietic cell transplantation: Change of paradigm. Viruses 2021, 13, 1530. [Google Scholar] [CrossRef]
- Hamdan, S.; Reddehase, M.J.; Holtappels, R. Cytomegalovirus immune evasion sets the functional avidity threshold for protection by CD8 T cells. Med. Microbiol. Immunol. 2022. [Google Scholar] [CrossRef]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human cytomegalovirus latency and reactivation in allogeneic hematopoietic stem cell transplant recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [Green Version]
- Quabeck, K. The lung as a critical organ in marrow transplantation. Bone Marrow Transplant. 1994, 14, S19–S28. [Google Scholar]
- Riddell, S.R. Pathogenesis of cytomegalovirus pneumonia in immunocompromised hosts. Semin. Respir. Infect. 1995, 10, 199–208. [Google Scholar]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Hematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 337–353. [Google Scholar]
- Michel, D.; Chevillotte, M.; Mertens, T. Antiviral therapy, drug resistance and computed resistance profiling. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 402–423. [Google Scholar]
- Chemaly, R.F.; Chou, S.; Einsele, H.; Griffiths, P.; Avery, R.; Razonable, R.R.; Mullane, K.M.; Kotton, C.; Lundgren, J.; Komatsu, T.E.; et al. Definitions of resistant and refractory cytomegalovirus infection and disease in transplant recipients for use in clinical trials. Clin. Infect. Dis. 2019, 68, 1420–1426. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Weiland, F.; Münch, K.; Jonjic, S.; Lüske, A.; Koszinowski, U.H. Interstitial murine cytomegalovirus pneumonia after irradiation: Characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 1985, 55, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, R.; Böhm, V.; Podlech, J.; Reddehase, M.J. CD8 T-cell based immunotherapy of cytomegalovirus infection: “proof of concept” provided by the murine model. Med. Microbiol. Immunol. 2008, 197, 125–134. [Google Scholar] [CrossRef]
- Ebert, S.; Podlech, J.; Gillert-Marien, D.; Gergely, K.M.; Büttner, J.K.; Fink, A.; Freitag, K.; Thomas, D.; Reddehase, M.J.; Holtappels, R. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Löffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mothy, M.; Or, R.; Mashan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Cowan, M.J.; Dunham, K.; Horn, B.; McGuirk, J.; Gilman, A.; Lucas, K.G. Adoptive immunotherapy with CMV-specific cytotoxic T lymphocytes for stem cell transplant patients with refractory CMV infections. J. Immunother. 2012, 35, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Odendahl, M.; Grigoleit, G.U.; Bönig, H.; Neuenhahn, M.; Albrecht, J.; Anderl, F.; Germeroth, L.; Schmitz, M.; Bornhäuser, M.; Einsele, H.; et al. Clinical-scale isolation of ‘minimally manipulated’ cytomegalovirus-specific donor lymphocytes for the treatment of refractory cytomegalovirus disease. Cytotherapy 2014, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.Y.; Zhao, X.Y.; Chang, Y.J.; Liu, J.; Xu, L.P.; Wang, Y.; Zhang, X.H.; Han, W.; Chen, Y.H.; Huang, X.J. Cytomegalovirus-specific T-cell transfer for refractory cytomegalovirus infection after haploidentical stem cell transplantation: The quantitative and qualitative immune recovery for cytomegalovirus. J. Infect. Dis. 2017, 216, 945–956. [Google Scholar] [CrossRef]
- Kaeuferle, T.; Krauss, R.; Blaeschke, F.; Willier, S.; Feuchtinger, T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Pahl-Seibert, M.F.; Juelch, M.; Podlech, J.; Thomas, D.; Deegen, P.; Reddehase, M.J.; Holtappels, R. Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J. Virol. 2005, 79, 5400–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, V.; Podlech, J.; Thomas, D.; Deegen, P.; Pahl-Seibert, M.F.; Lemmermann, N.A.; Grzimek, N.K.; Oehrlein-Karpi, S.A.; Reddehase, M.J.; Holtappels, R. Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med. Microbiol. Immunol. 2008, 197, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Klobuch, S.; Podlech, J.; Plachter, B.; Hoffmann, P.; Renzaho, A.; Theobald, M.; Reddehase, M.J.; Herr, W.; Lemmermann, N.A. Evaluating human T-cell therapy of cytomegalovirus organ disease in HLA-transgenic mice. PLoS Pathog. 2015, 11, e1005049. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, M.; Khan, N.; Pourgheysari, B.; Tauro, S.; McDonald, D.; Osman, H.; Assenmacher, M.; Billingham, L.; Steward, C.; Crawley, C.; et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J. Exp. Med. 2005, 202, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Lemmermann, N.A.; Podlech, J.; Seckert, C.K.; Kropp, K.A.; Grzimek, N.K.; Reddehase, M.J.; Holtappels, R. CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In Methods in Microbiology: Immunology of Infection, 3rd ed.; Kabelitz, D., Kaufmann, S.H.E., Eds.; Academic Press: London, UK, 2010; Volume 37, pp. 369–420. [Google Scholar]
- Gergely, K.M.; Podlech, J.; Becker, S.; Freitag, K.; Krauter, S.; Büscher, N.; Holtappels, R.; Plachter, B.; Reddehase, M.J.; Lemmermann, N.A.W. Therapeutic vaccination of hematopoietic cell transplantation recipients improves protective CD8 T-cell immunotherapy of cytomegalovirus infection. Front. Immunol. 2021, 12, 694588. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Krmpotic, A.; Podlech, J.; Brizic, I.; Prager, A.; Adler, H.; Karbach, A.; Wu, Y.; Jonjic, S.; Reddehase, M.J.; et al. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog. 2015, 11, e1004640. [Google Scholar] [CrossRef]
- Podlech, J.; Reddehase, M.J.; Adler, B.; Lemmermann, N.A. Principles for studying in vivo attenuation of virus mutants: Defining the role of the cytomegalovirus gH/gL/gO complex as a paradigm. Med. Microbiol. Immunol. 2015, 204, 295–305. [Google Scholar] [CrossRef]
- Holtappels, R.; Freitag, K.; Renzaho, A.; Becker, S.; Lemmermann, N.A.W.; Reddehase, M.J. Revisiting CD8 T-cell ‘memory inflation’: New insights with implications for cytomegaloviruses as vaccine vectors. Vaccines 2020, 8, 402. [Google Scholar] [CrossRef]
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef]
- Welten, S.P.M.; Baumann, N.S.; Oxenius, A. Fuel and brake of memory T cell inflation. Med. Microbiol. Immunol. 2019, 208, 329–338. [Google Scholar] [CrossRef]
- Cicin-Sain, L. Cytomegalovirus memory inflation and immune protection. Med. Microbiol. Immunol. 2019, 208, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Munks, M.W.; Gold, M.C.M.; Zajac, A.L.; Doom, C.M.; Morello, C.S.; Spector, D.H.; Hill, A.B. Genome-wide analysis reveals a highly diverse CD8 T cell response to murine cytomegalovirus. J. Immunol. 2006, 176, 3760–3766. [Google Scholar] [CrossRef] [PubMed]
- Seckert, C.K.; Schader, S.I.; Ebert, S.; Thomas, D.; Freitag, K.; Renzaho, A.; Podlech, J.; Reddehase, M.J.; Holtappels, R. Antigen-presenting cells of haematopoietic origin prime cytomegalovirus-specific CD8 T-cells but are not sufficient for driving memory inflation during viral latency. J. Gen. Virol. 2011, 92, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Thomas, D.; Podlech, J.; Reddehase, M.J. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J. Virol. 2002, 76, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Kropp, K.A.; Simon, C.O.; Fink, A.; Renzaho, A.; Kühnapfel, B.; Podlech, J.; Reddehase, M.J.; Grzimek, N.K.A. Synergism between the components of the bipartite major immediate-early transcriptional enhancer of murine cytomegalovirus does not accelerate virus replication in cell culture and host tissues. J. Gen. Virol. 2009, 90, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Stemberger, C.; Graef, P.; Odendahl, M.; Albrecht, J.; Dössinger, G.; Anderl, F.; Buchholz, V.R.; Gasteiger, G.; Schiemann, M.; Grigoleit, G.U.; et al. Lowest numbers of primary CD8(+) T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood 2014, 124, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Celis, J.E.; Madsen, P.; Celis, A.; Nielsen, H.V.; Gesser, B. Cyclin (PCNA, auxiliary protein of DNA polymerase delta) is a central component of the pathway(s) leading to DNA replication and cell division. FEBS Lett. 1987, 220, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kurki, P.; Ogata, K.; Tan, E.M. Monoclonal antibodies to proliferating cell nuclear antigen (PCNA)/cyclin as probes for proliferating cells by immunofluorescence microscopy and flow cytometry. J. Immunol. Methods 1988, 109, 49–59. [Google Scholar] [CrossRef]
- Bologna-Molina, R.; Mosqueda-Taylor, A.; Molina-Frechero, N.; Mori-Estevez, A.D.; Sánchez-Acuña, G. Comparison of the value of PCNA and Ki-67 as markers of cell proliferation in ameloblastic tumors. Med. Oral Patol. Oral Y Cir. Buccal 2013, 18, e174–e179. [Google Scholar] [CrossRef]
- Hall, P.A.; Levison, D.A.; Woods, A.L.; Yu, C.C.; Kellock, D.B.; Watkins, J.A.; Barnes, D.M.; Gillet, C.E.; Camplejohn, R.; Waseem, N.H.; et al. Proliferating cell nuclear antigen (PCNA) immunolocalization in paraffin sections: An index of cell proliferation with evidence of deregulated expression in some neoplasms. J. Path. 1990, 162, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Mate, J.L.; Ariza, A.; Muñoz, A.; Molinero, J.L.; López, D.; Navas-Palacios, J.J. Induction of proliferating cell nuclear antigen and Ki-67 expression by cytomegalovirus infection. J. Pathol. 1998, 184, 279–282. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holtappels, R.; Podlech, J.; Freitag, K.; Lemmermann, N.A.; Reddehase, M.J. Memory CD8 T Cells Protect against Cytomegalovirus Disease by Formation of Nodular Inflammatory Foci Preventing Intra-Tissue Virus Spread. Viruses 2022, 14, 1145. https://doi.org/10.3390/v14061145
Holtappels R, Podlech J, Freitag K, Lemmermann NA, Reddehase MJ. Memory CD8 T Cells Protect against Cytomegalovirus Disease by Formation of Nodular Inflammatory Foci Preventing Intra-Tissue Virus Spread. Viruses. 2022; 14(6):1145. https://doi.org/10.3390/v14061145
Chicago/Turabian StyleHoltappels, Rafaela, Jürgen Podlech, Kirsten Freitag, Niels A. Lemmermann, and Matthias J. Reddehase. 2022. "Memory CD8 T Cells Protect against Cytomegalovirus Disease by Formation of Nodular Inflammatory Foci Preventing Intra-Tissue Virus Spread" Viruses 14, no. 6: 1145. https://doi.org/10.3390/v14061145