
Evolutionary Dynamics of Mexican Lineage H5N2 Avian Influenza Viruses

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation and Genome Sequencing
2.2. Nucleotide Sequences Used in the Study
2.3. Sequence Analyses
2.4. Intravenous Pathogenicity Index of 2016–2019 Mexican H5N2 AIVs
2.5. Analysis of Selection Pressure
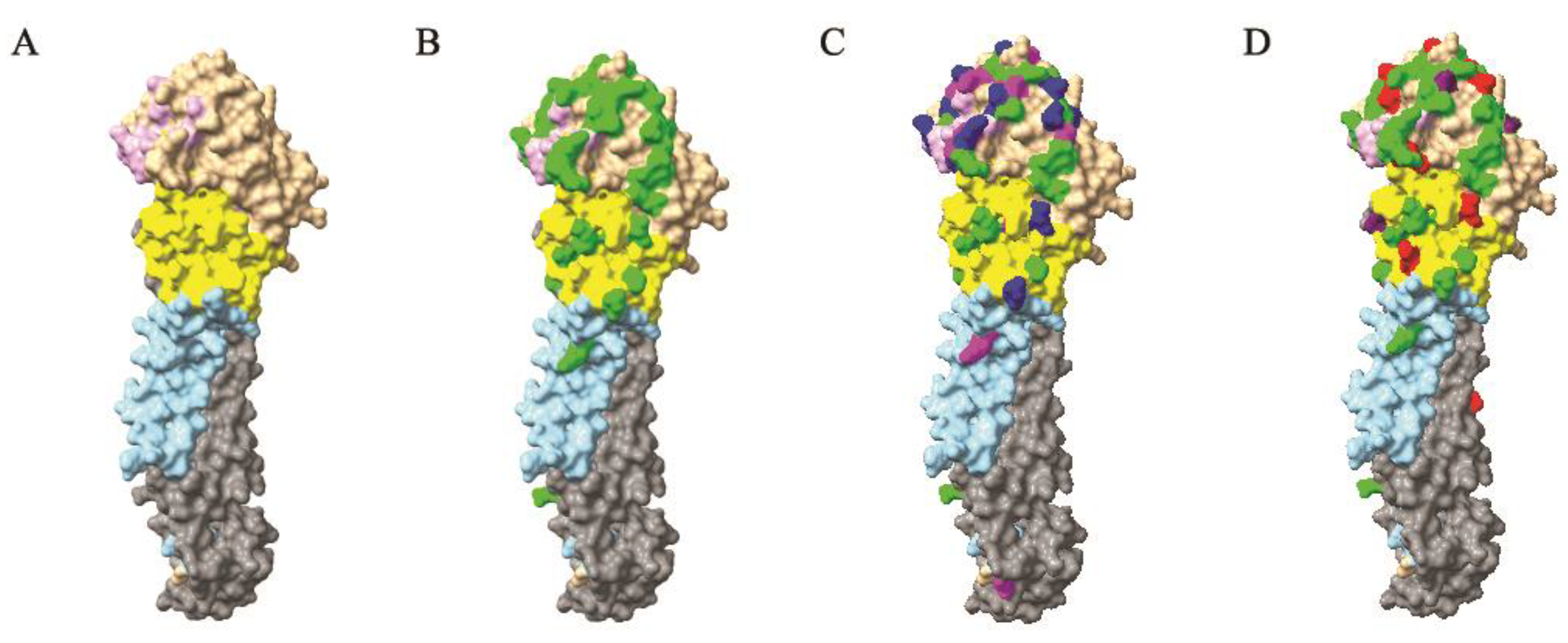
2.6. 3D Structural Analyses
3. Results
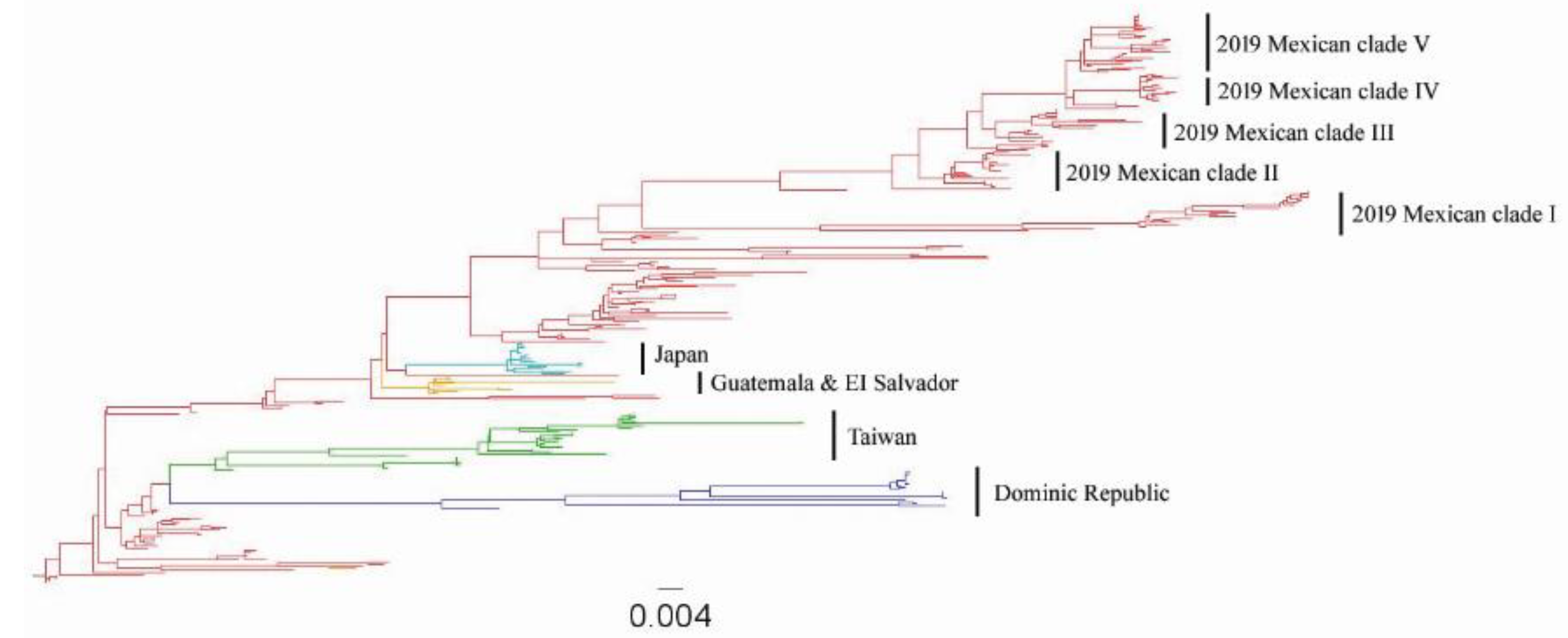
3.1. Origins of the Mexican Lineage H5N2 AIV
3.2. The Evolutionary Rate of Mexican Lineage H5N2 AIV
3.3. Protein-Level Evolution and Selection Analysis of Mexican Lineage H5N2 AIVs
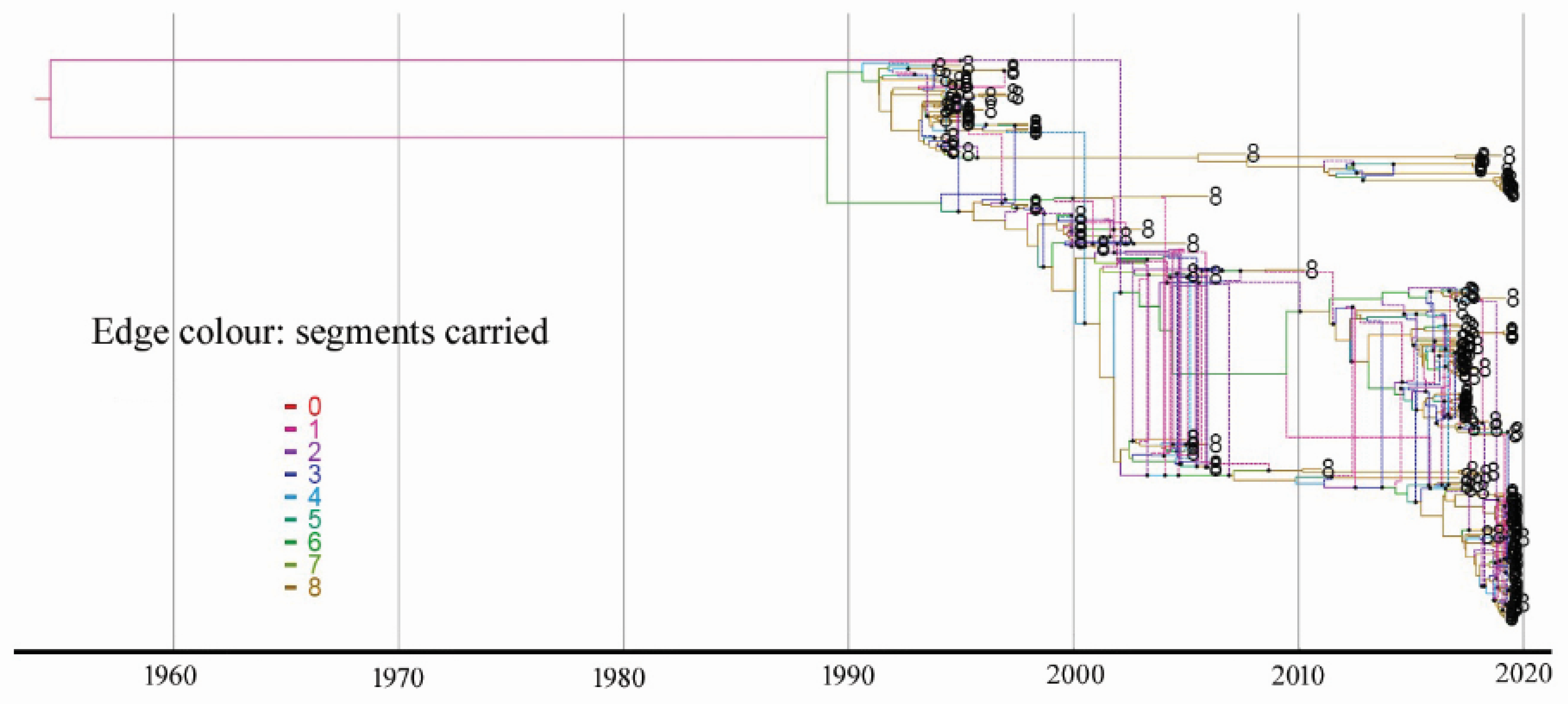
3.4. Estimate of Reassortment
3.5. Diversity of the Mexican Lineage H5N2 AIV and HA Cleavage Sites
3.6. Antigenic Evolution of H5N2 AIVs in Mexico
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martinez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouchier, R.A.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A.D. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D. Avian Influenza; Office International des Epizooties: Paris, France, 2021. [Google Scholar]
- Swayne, D.E.; Sims, L.D. Influenza. In Diseases of Poultry, 14th ed.; Swayne, D.E., Boulianne, M., Logue, C.M., McDougald, L.R., Nair, V., Suarez, D.L., Wit, S., Grimes, T., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; pp. 210–256. [Google Scholar]
- Horimoto, T.; Rivera, E.; Pearson, J.; Senne, D.; Krauss, S.; Kawaoka, Y.; Webster, R.G. Origin and molecular changes associated with emergence of a highly pathogenic H5N2 influenza virus in Mexico. Virology 1995, 213, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.; Crawford, J.M.; Latimer, J.W.; Rivera-Cruz, E.; Perdue, M.L. Heterogeneity in the haemagglutinin gene and emergence of the highly pathogenic phenotype among recent H5N2 avian influenza viruses from Mexico. J. Gen. Virol. 1996, 77, 1493–1504. [Google Scholar] [CrossRef]
- Bertran, K.; Criado, M.F.; Lee, D.H.; Killmaster, L.; Sa, E.; Silva, M.; Lucio, E.; Widener, J.; Pritchard, N.; Atkins, E.; et al. Protection of white leghorn chickens by recombinant fowlpox vector vaccine with an updated H5 insert against Mexican H5N2 avian influenza viruses. Vaccine 2020, 38, 1526–1534. [Google Scholar]
- Suarez, D.L.; Spackman, E.; Senne, D.A. Update on molecular epidemiology of H1, H5, and H7 influenza virus infections in poultry in North America. Avian Dis. 2003, 47 (Suppl. 3), 888–897. [Google Scholar] [CrossRef]
- Lee, C.C.; Zhu, H.; Huang, P.Y.; Peng, L.; Chang, Y.C.; Yip, C.H.; Li, Y.T.; Cheung, C.L.; Compans, R.; Yang, C.; et al. Emergence and evolution of avian H5N2 influenza viruses in chickens in Taiwan. J. Virol. 2014, 88, 5677–5686. [Google Scholar] [CrossRef] [Green Version]
- Okamatsu, M.; Saito, T.; Yamamoto, Y.; Mase, M.; Tsuduku, S.; Nakamura, K.; Tsukamoto, K.; Yamaguchi, S. Low pathogenicity H5N2 avian influenza outbreak in Japan during the 2005–2006. Vet. Microbiol. 2007, 124, 35–46. [Google Scholar] [CrossRef]
- Chung, D.H.; Gomez, D.R.; Vargas, J.M.; Amador, B.L.; Torchetti, M.K.; Killian, M.L.; Swayne, D.E.; Lee, D.H. Low pathogenicity avian influenza (H5N2) viruses, Dominican Republic. Emerg. Infect. Dis. 2020, 26, 3094–3096. [Google Scholar] [CrossRef]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Muller, N.F.; Stolz, U.; Dudas, G.; Stadler, T.; Vaughan, T.G. Bayesian inference of reassortment networks reveals fitness benefits of reassortment in human influenza viruses. Proc. Natl. Acad. Sci. USA 2020, 117, 17104–17111. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained Bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, J.; Blixt, O.; Tumpey, T.M.; Taubenberger, J.K.; Paulson, J.C.; Wilson, I.A. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 2006, 312, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Suzuki, Y.; Suzuki, T.; Le, M.Q.; Nidom, C.A.; Sakai-Tagawa, Y.; Muramoto, Y.; Ito, M.; Kiso, M.; Horimoto, T.; et al. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 2006, 444, 378–382. [Google Scholar] [CrossRef]
- Yen, H.L.; Aldridge, J.R.; Boon, A.C.; Ilyushina, N.A.; Salomon, R.; Hulse-Post, D.J.; Marjuki, H.; Franks, J.; Boltz, D.A.; Bush, D.; et al. Changes in H5N1 influenza virus hemagglutinin receptor binding domain affect systemic spread. Proc. Natl. Acad. Sci. USA 2009, 106, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.M.; Blixt, O.; Stevens, J.; Lipatov, A.S.; Davis, C.T.; Collins, B.E.; Cox, N.J.; Paulson, J.C.; Donis, R.O. In vitro evolution of H5N1 avian influenza virus toward human-type receptor specificity. Virology 2012, 422, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Senne, D.A.; Suarez, D.L. Effect of vaccine use in the evolution of Mexican lineage H5N2 avian influenza virus. J. Virol. 2004, 78, 8372–8381. [Google Scholar] [CrossRef] [Green Version]
- Escorcia, M.; Vazquez, L.; Mendez, S.T.; Rodriguez-Ropon, A.; Lucio, E.; Nava, G.M. Avian influenza: Genetic evolution under vaccination pressure. Virol. J. 2008, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Escorcia, M.; Carrillo-Sanchez, K.; March-Mifsut, S.; Chapa, J.; Lucio, E.; Nava, G.M. Impact of antigenic and genetic drift on the serologic surveillance of H5N2 avian influenza viruses. BMC Vet. Res. 2010, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, H.L.; Lambrecht, B.; van Borm, S.; Torrieri-Dramard, L.; Klatzmann, D.; Bellier, B.; van den Berg, T. Identification of a dominant epitope in the hemagglutinin of an Asian highly pathogenic avian influenza H5N1 clade 1 virus by selection of escape mutants. Avian Dis. 2010, 54, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Kaverin, N.V.; Rudneva, I.A.; Ilyushina, N.A.; Varich, N.L.; Lipatov, A.S.; Smirnov, Y.A.; Govorkova, E.A.; Gitelman, A.K.; Lvov, D.K.; Webster, R.G. Structure of antigenic sites on the haemagglutinin molecule of H5 avian influenza virus and phenotypic variation of escape mutants. J. Gen. Virol. 2002, 83, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Kaverin, N.V.; Rudneva, I.A.; Govorkova, E.A.; Timofeeva, T.A.; Shilov, A.A.; Kochergin-Nikitsky, K.S.; Krylov, P.S.; Webster, R.G. Epitope mapping of the hemagglutinin molecule of a highly pathogenic H5N1 influenza virus by using monoclonal antibodies. J. Virol. 2007, 81, 12911–12917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudneva, I.A.; Kushch, A.A.; Masalova, O.V.; Timofeeva, T.A.; Klimova, R.R.; Shilov, A.A.; Ignatieva, A.V.; Krylov, P.S.; Kaverin, N.V. Antigenic epitopes in the hemagglutinin of Qinghai-type influenza H5N1 virus. Viral Immunol. 2010, 23, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Philpott, M.; Easterday, B.C.; Hinshaw, V.S. Neutralizing epitopes of the H5 hemagglutinin from a virulent avian influenza virus and their relationship to pathogenicity. J. Virol. 1989, 63, 3453–3458. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.L.; Chen, Y.; Wang, P.; Song, W.; Lau, S.Y.; Rayner, J.M.; Smith, G.J.; Webster, R.G.; Peiris, J.S.; Lin, T.; et al. Antigenic profile of avian H5N1 viruses in Asia from 2002 to 2007. J. Virol. 2008, 82, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- Prabakaran, M.; He, F.; Meng, T.; Madhan, S.; Yunrui, T.; Jia, Q.; Kwang, J. Neutralizing epitopes of influenza virus hemagglutinin: Target for the development of a universal vaccine against H5N1 lineages. J. Virol. 2010, 84, 11822–11830. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Paul, S.S.; Mo, X.; Yuan, Y.A.; Tan, Y.J. The vestigial esterase domain of haemagglutinin of H5N1 avian influenza A virus: Antigenicity and contribution to viral pathogenesis. Vaccines 2018, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Swayne, D.E. Pathobiology of H5N2 Mexican avian influenza virus infections of chickens. Vet. Pathol. 1997, 34, 557–567. [Google Scholar] [CrossRef]
- Giannecchini, S.; Clausi, V.; Di Trani, L.; Falcone, E.; Terregino, C.; Toffan, A.; Cilloni, F.; Matrosovich, M.; Gambaryan, A.S.; Bovin, N.V.; et al. Molecular adaptation of an H7N3 wild duck influenza virus following experimental multiple passages in quail and turkey. Virology 2010, 408, 167–173. [Google Scholar] [CrossRef]
- Li, J.; zu Dohna, H.; Anchell, N.L.; Adams, S.C.; Dao, N.T.; Xing, Z.; Cardona, C.J. Adaptation and transmission of a duck-origin avian influenza virus in poultry species. Virus Res. 2010, 147, 40–46. [Google Scholar] [CrossRef]
- Bataille, A.; van der Meer, F.; Stegeman, A.; Koch, G. Evolutionary analysis of inter-farm transmission dynamics in a highly pathogenic avian influenza epidemic. PLoS Pathog. 2011, 7, e1002094. [Google Scholar] [CrossRef] [Green Version]
- Monne, I.; Fusaro, A.; Nelson, M.I.; Bonfanti, L.; Mulatti, P.; Hughes, J.; Murcia, P.R.; Schivo, A.; Valastro, V.; Moreno, A.; et al. Emergence of a highly pathogenic avian influenza virus from a low-pathogenic progenitor. J. Virol. 2014, 88, 4375–4388. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Berhane, Y.; Dube, C.; Liang, B.; Pasick, J.; VanDomselaar, G.; Alexandersen, S. Epidemiological and evolutionary inference of the transmission Network of the 2014 highly pathogenic avian influenza H5N2 outbreak in British Columbia, Canada. Sci. Rep. 2016, 6, 30858. [Google Scholar] [CrossRef] [Green Version]
- Kamal, R.P.; Kumar, A.; Davis, C.T.; Tzeng, W.P.; Nguyen, T.; Donis, R.O.; Katz, J.M.; York, I.A. Emergence of highly pathogenic avian influenza A(H5N1) virus PB1-F2 variants and their virulence in BALB/c Mice. J. Virol. 2015, 89, 5835–5846. [Google Scholar] [CrossRef] [Green Version]
- Sealy, J.E.; Yaqub, T.; Peacock, T.P.; Chang, P.; Ermetal, B.; Clements, A.; Sadeyen, J.R.; Mehboob, A.; Shelton, H.; Bryant, J.E.; et al. Association of increased receptor-binding avidity of influenza A(H9N2) viruses with escape from antibody-based immunity and enhanced zoonotic potential. Emerg. Infect. Dis. 2018, 25, 63–72. [Google Scholar] [CrossRef]
- Nelson, M.I.; Viboud, C.; Simonsen, L.; Bennett, R.T.; Griesemer, S.B.; George, K.S.; Taylor, J.; Spiro, D.J.; Sengamalay, N.A.; Ghedin, E.; et al. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog. 2008, 4, e1000012. [Google Scholar] [CrossRef] [Green Version]
- Murcia, P.R.; Wood, J.L.; Holmes, E.C. Genome-scale evolution and phylodynamics of equine H3N8 influenza A virus. J. Virol. 2011, 85, 5312–5322. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Hughes, J.; Murcia, P.R. Origins and evolutionary dynamics of H3N2 canine influenza virus. J. Virol. 2015, 89, 5406–5418. [Google Scholar] [CrossRef] [Green Version]
- Trovao, N.S.; Talavera, G.A.; Nelson, M.I.; Perez de la Rosa, J.D. Evolution of highly pathogenic H7N3 avian influenza viruses in Mexico. Zoonoses Public Health 2020, 67, 318–323. [Google Scholar] [CrossRef]
- Youk, S.; Lee, D.H.; Ferreira, H.L.; Afonso, C.L.; Absalon, A.E.; Swayne, D.E.; Suarez, D.L.; Pantin-Jackwood, M.J. Rapid Evolution of Mexican H7N3 highly pathogenic avian influenza viruses in poultry. PLoS ONE 2019, 14, e0222457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abolnik, C. Evolution of H5 highly pathogenic avian influenza: Sequence data indicate stepwise changes in the cleavage site. Arch. Virol. 2017, 162, 2219–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laleye, A.T.; Abolnik, C. Emergence of highly pathogenic H5N2 and H7N1 influenza A viruses from low pathogenic precursors by serial passage in Ovo. PLoS ONE 2020, 15, e0240290. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Lee, R.T.; Gunalan, V.; Eisenhaber, F. The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA. Virol. J. 2013, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Nao, N.; Yamagishi, J.; Miyamoto, H.; Igarashi, M.; Manzoor, R.; Ohnuma, A.; Tsuda, Y.; Furuyama, W.; Shigeno, A.; Kajihara, M.; et al. Genetic predisposition to acquire a polybasic cleavage site for highly pathogenic avian influenza virus hemagglutinin. mBio 2017, 8, e02298-16. [Google Scholar] [CrossRef] [Green Version]
- Pasick, J.; Handel, K.; Robinson, J.; Copps, J.; Ridd, D.; Hills, K.; Kehler, H.; Cottam-Birt, C.; Neufeld, J.; Berhane, Y.; et al. Intersegmental recombination between the haemagglutinin and matrix genes was responsible for the emergence of a highly pathogenic H7N3 avian influenza virus in British Columbia. J. Gen. Virol. 2005, 86, 727–731. [Google Scholar] [CrossRef]
- Suarez, D.L.; Senne, D.A.; Banks, J.; Brown, I.H.; Essen, S.C.; Lee, C.W.; Manvell, R.J.; Mathieu-Benson, C.; Moreno, V.; Pedersen, J.C.; et al. Recombination resulting in virulence shift in avian influenza outbreak, Chile. Emerg. Infect. Dis. 2004, 10, 693–699. [Google Scholar] [CrossRef]
- Abolnik, C.; Olivier, A.; Reynolds, C.; Henry, D.; Cumming, G.; Rauff, D.; Romito, M.; Petty, D.; Falch, C. Susceptibility and status of avian influenza in ostriches. Avian Dis. 2016, 60, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, C. Experience in control of avian influenza in the Americas. Dev. Biol. 2007, 130, 53–60. [Google Scholar]
- Perdue, M.L.; Suarez, D.L. Structural features of the avian influenza virus hemagglutinin that influence virulence. Vet. Microbiol. 2000, 74, 77–86. [Google Scholar] [CrossRef]
- Baigent, S.J.; McCauley, J.W. Glycosylation of haemagglutinin and stalk-length of neuraminidase combine to regulate the growth of avian influenza viruses in tissue culture. Virus Res. 2001, 79, 177–185. [Google Scholar] [CrossRef]
- Swayne, D.E.; Pavade, G.; Hamilton, K.; Vallat, B.; Miyagishima, K. Assessment of national strategies for control of high-pathogenicity avian influenza and low-pathogenicity notifiable avian influenza in poultry, with emphasis on vaccines and vaccination. Rev. Sci. Tech. 2011, 30, 839–870. [Google Scholar] [CrossRef] [PubMed]
- Swayne, D.E.; Beck, J.R.; Garcia, M.; Stone, H.D. Influence of virus strain and antigen mass on efficacy of H5 avian influenza inactivated vaccines. Avian Pathol. 1999, 28, 245–255. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Substitution Rate (Subs/Site/Year) | tMRCA (Year) | ||
|---|---|---|---|---|
| Mean (×10−3) | 95% HPD (×10−3) | Mean | 95% HPD (×10−3) | |
| PB2 | 4.32 | 4.01–4.64 | 1990.80 | 1988.88–1992.34 |
| PB1 | 3.79 | 3.56–4.04 | 1991.15 | 1989.03–1992.62 |
| PA | 3.80 | 3.52–4.06 | 1990.75 | 1988.30–1992.66 |
| HA | 5.69 | 5.32–6.06 | 1990.27 | 1987.92–1992.21 |
| NP | 3.78 | 3.50–4.08 | 1990.38 | 1987.08–1992.54 |
| NA | 5.49 | 5.09–5.91 | 1991.35 | 1988.04–1993.22 |
| MP | 3.59 | 3.27–3.89 | 1990.33 | 1987.43–1992.69 |
| NS | 4.43 | 4.10–4.76 | 1990.97 | 1988.83–1992.73 |
| Gene | Codon Position | SLAC | FUBAR | MEME | |||
|---|---|---|---|---|---|---|---|
| dN-dS | p-Value | dN-dS | Posterior Probability | p-Value | Number of Branches under Episodic Selection | ||
| PB2 | 191 | 1.68 | 0.90 | ||||
| PB1 | 296 | 0.00 | 1 | ||||
| 746 | 2.49 | 0.09 | 1.59 | 0.91 | |||
| 757 | 0.00 | 1 | |||||
| PA | 210 | 3.56 | 0.08 | ||||
| 489 | 5.02 | 0.05 | 5.91 | 0.98 | 0.00 | 12 | |
| PA-X | 193 | 3.48 | 0.08 | 2.11 | 0.98 | ||
| 216 | 3.32 | 0.06 | 1.91 | 0.97 | |||
| 218 | 2.85 | 0.09 | 1.57 | 0.95 | |||
| 221 | 2.74 | 0.09 | |||||
| H5 | 88 (72) * | 2.44 | 0.06 | 2.06 | 0.97 | ||
| 139 (123) | 3.25 | 0.01 | 2.19 | 0.99 | 0.00 | 9 | |
| 153 (137) | 5.09 | 0.00 | 6.33 | 1.00 | 0.00 | 5 | |
| 171 (155) | 4.41 | 0.00 | 3.59 | 0.99 | 0.00 | 7 | |
| 174 (158) | 0.00 | 11 | |||||
| 194 (178) | 2.54 | 0.07 | 2.00 | 0.95 | |||
| 197 (181) | 2.96 | 0.04 | 1.94 | 0.95 | |||
| 201 (185) | 0.00 | 2 | |||||
| 202 (186) | 0.00 | 4 | |||||
| 210 (194) | 4.96 | 0.01 | 5.20 | 0.97 | 0.00 | 5 | |
| 286 (270) | 2.16 | 0.08 | 1.44 | 0.92 | |||
| 392 (376) | 2.43 | 0.06 | 1.91 | 0.96 | |||
| NP | 452 | 3.44 | 0.07 | 2.47 | 0.95 | ||
| N2 | 43 | 2.61 | 0.08 | 1.75 | 0.97 | ||
| 58 | 3.55 | 0.06 | |||||
| 362 | 0.00 | 1 | |||||
| 449 | 0.00 | 3 | |||||
| M2 | 13 | 2.49 | 0.98 | ||||
| 20 | 1.63 | 0.94 | |||||
| 28 | 4.81 | 0.95 | |||||
| 89 | 1.83 | 0.94 | |||||
| NS1 | 63 | 3.04 | 0.06 | 2.57 | 0.97 | ||
| 64 | 1.72 | 0.92 | |||||
| 176 | 2.20 | 0.93 | |||||
| 205 | 1.81 | 0.94 | |||||
| 210 | 2.60 | 0.08 | 2.11 | 0.96 | |||
| 215 | 4.24 | 0.02 | 4.94 | 0.98 | 0.01 | 6 | |
| 221 | 2.68 | 0.95 | |||||
| 226 | 2.17 | 0.94 | |||||
| NS2 | 14 | 3.58 | 0.96 | ||||
| 44 | 2.74 | 0.92 | |||||
| Amino Acid Position a | Structural Location b | Sequons c | Percentage of Isolates (%) |
|---|---|---|---|
| 10 | stalk | NNST | 99.5 |
| 23 | stalk | NVTV | 99.5 |
| 45 | VED | NGTK | 1.9 |
| 54 | VED | NCSV | 2.4 |
| 72 | VED | NVSE | 1.4 |
| 84 | VED | NPSN | 53.3 |
| 126 | RBD | NASA | 5.7 |
| NASS | 67.1 | ||
| 138 | RBD | NGSS | 0.5 |
| 161 | RBD | NRTY | 0.5 |
| 163 | RBD | NYTN | 81.9 |
| 165 | RBD | NNTN | 14.3 |
| 193 | RBD | NPST | 1.0 |
| NPTT | 0.5 | ||
| 236 | RBD | NDSI | 99.5 |
| 275 | stalk | NATC | 1.4 |
| 286 | stalk | NSSL | 79.5 |
| NSSM | 14.3 | ||
| NTSM | 2.9 | ||
| 319 | stalk | NVSQ | 0.5 |
| 480 (484) | stalk | NGTY | 100.0 |
| 539 (543) | CD | NGSL | 100.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.; Navarro-López, R.; Solis-Hernandez, M.; Liljehult-Fuentes, F.; Molina-Montiel, M.; Lagunas-Ayala, M.; Rocha-Martinez, M.; Ferrara-Tijera, E.; Pérez de la Rosa, J.; Berhane, Y. Evolutionary Dynamics of Mexican Lineage H5N2 Avian Influenza Viruses. Viruses 2022, 14, 958. https://doi.org/10.3390/v14050958
Xu W, Navarro-López R, Solis-Hernandez M, Liljehult-Fuentes F, Molina-Montiel M, Lagunas-Ayala M, Rocha-Martinez M, Ferrara-Tijera E, Pérez de la Rosa J, Berhane Y. Evolutionary Dynamics of Mexican Lineage H5N2 Avian Influenza Viruses. Viruses. 2022; 14(5):958. https://doi.org/10.3390/v14050958
Chicago/Turabian StyleXu, Wanhong, Roberto Navarro-López, Mario Solis-Hernandez, Francisco Liljehult-Fuentes, Miguel Molina-Montiel, María Lagunas-Ayala, Marisol Rocha-Martinez, Eduardo Ferrara-Tijera, Juan Pérez de la Rosa, and Yohannes Berhane. 2022. "Evolutionary Dynamics of Mexican Lineage H5N2 Avian Influenza Viruses" Viruses 14, no. 5: 958. https://doi.org/10.3390/v14050958
APA StyleXu, W., Navarro-López, R., Solis-Hernandez, M., Liljehult-Fuentes, F., Molina-Montiel, M., Lagunas-Ayala, M., Rocha-Martinez, M., Ferrara-Tijera, E., Pérez de la Rosa, J., & Berhane, Y. (2022). Evolutionary Dynamics of Mexican Lineage H5N2 Avian Influenza Viruses. Viruses, 14(5), 958. https://doi.org/10.3390/v14050958