Abstract
As nucleocytoplasmic large DNA viruses, replication of ranaviruses (genus Ranavirus, family Iridoviridae) involves a series of viral and host proteins. We have described that the replication and transcription machinery of Andrias davidianus ranavirus (ADRV) which was isolated from the Chinese giant salamander contained host factors. Here, a new host factor, the MutS homolog 2 (MSH2), was proved as an important protein that participated in ADRV infection. Expression of MSH2 was stable during ADRV infection in cultured cells and it localized at the cytoplasmic viral factories and colocalized with virus nascent DNA, indicating its possible role in virus genome replication. Investigation of the viral proteins that interacted with MSH2 by co-immunoprecipitation showed that A. davidianus MSH2 can interact with ADRV-35L (possible components associated with virus transcription), ADRV-47L (virus DNA polymerase), and ADRV-98R. Further knockdown MSH2 expression by RNAi significantly reduced the late gene expression of ADRV. Additionally, MSH2 knockout by CRISPR/Cas9 significantly reduced viral titers, genome replication, and late gene transcription of ADRV. Thus, the current study proved that ADRV can engage cellular MSH2 for its efficient genome replication and late gene transcription, which provided new information for understanding the roles of host factors in ranavirus replication and transcription.
1. Introduction
Ranaviruses are nucleocytoplasmic large DNA viruses that belong to the family Iridoviridae [1,2], which have been isolated from aquatic animals globally, such as bony fish, amphibians, and reptiles [3,4,5,6]. Because of the wide host range, ranaviruses represent a great threat to cultured and wild aquatic animals [7,8,9]. The sequenced ranaviruses possessed genomes with a unit size of 104–140 kbp, which can encode more than 100 proteins [1].
Ranavirus genome replication has been reported to occur in two-stages involving the nucleus and cytoplasm [10]. Although several viral proteins involved in ranavirus DNA replication have been predicted or investigated [11,12,13], the mechanism of ranavirus DNA replication still needs to be explored. Andrias davidianus ranavirus (ADRV) was isolated from diseased Chinese giant salamanders, which have a genome size of 106.7 kbp with 101 predicted open reading frames (ORFs) [6]. Because of the intracellular life cycle, virus replication involves not only viral proteins but also host proteins. In a recent study on the replication and transcription machinery of ranaviruses, we identified several viral and host proteins as potential proteins associated with viral nascent DNA by proteomic analysis of ADRV infected cells [14]. Among them, the host MutS homolog 2 (MSH2) was found but its function in virus infection was unknown.
MSH2 belonged to the DNA mismatch repair (MMR) proteins, which are highly conserved from lower to higher animals [15]. MSH2 along with the other MMR proteins such as MSH3 and MSH6 play important functions in maintaining the integrity of the genome in normal cells [16,17]. They can recognize the mismatches and insertion/deletion loops that occur in the genome and thus are needed for the repair of DNA replication errors [18]. The MMR proteins have been shown to be involved in the replication of the herpes viruses including herpes simplex virus 1 and Epstein-Barr virus [19,20], which are DNA viruses replicated in the nucleus. However, the function of DNA mismatch repair protein in ranavirus replication which mainly occurred in the cytoplasm is unknown.
In the present study, we explored the function of MSH2 in ADRV infection and replication by immunofluorescence, co-immunoprecipitation, RNA interference, and CRISPR/Cas9 knockout. The results showed that MSH2 can interact with virus proteins and localize in viral factories. Knockdown or knockout of MSH2 expression reduced virus genome replication and late gene expression.
2. Materials and Methods
2.1. Virus and Cells
ADRV and RGV (Rana grylio virus) that were maintained in our lab were used in the present study [6,21]. The Chinese giant salamander thymus cell (GSTC) line was the culture in the M199 medium supplemented with 10% fetal bovine serum at 25 °C until use [22]. Baby hamster kidney fibroblast cells (BHK-21) and human embryonic kidney (HEK293T) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2 [14]. To prepare virus stocks, GSTC cells were infected with ADRV or RGV at an MOI of 0.1. Three days post-infection, the infected cells were collected, freeze-thawed, aliquoted, and kept at −80 °C until use.
2.2. MSH2 Gene Cloning and Plasmid Construction
Total RNA was extracted from GSTC cells with TRIzol Reagent (Thermo Fisher, Waltham, MA, USA) and the first strand of cDNA was synthesized with HiScript III 1st Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme, Nanjing, China) according to the manufacturer’s recommendations. The coding sequence of the MSH2 gene was amplified with primers (5′-AGCCTGATGCTGATGT CTAGAATGGCGGTGCAACCC-3′/5′-ACCCTGAAGTTCTCAGGATCCTTATGCAGTAGTCTTTGTTCG-3′) by PCR based on the sequences from previous transcriptomic data [23] and then ligated into the pCGN-HAM vector by infusion cloning to generate 3HA-tagged MSH2 protein. The recombinant plasmids expressing 3Flag-tagged viral proteins were constructed as described previously [14]. The obtained plasmids were proved by DNA sequencing.
2.3. Western Blotting
BHK-21 cells were infected with ADRV at an MOI of 0.5. The cells were harvested at the indicated time points and subjected to Western blotting analysis as described previously [24]. A rabbit anti-MSH2 antibody (A8740, ABclonal, Wuhan, China) was used as the primary antibody, and the corresponding horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (ABclonal, Wuhan, China) was used as the secondary antibody. Antibody binding was detected by chemiluminescence (Millipore, Burlington, MA, USA). Detection of β-actin was used as an internal control.
2.4. EdU Labeling and Immunofluorescence
EdU labeling was performed as described previously [14]. Briefly, BHK-21 cells plated on coverslips were infected with ADRV or RGV at an MOI of 0.5 and incubated at 28 °C. EdU (Invitrogen) was added to a final concentration of 10 μM at the times indicated. After continued incubation for 30 min, the cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde, and then processed with a Click-iT Plus EdU Cell Proliferation Kit for Imaging (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocols. Alexa Fluor 488-azide was used in the kit to label the EdU. In the following immunofluorescence assay, the EdU labeled cells were then incubated with antibodies against MSH2 (rabbit anti-MSH2 antibody, ABclonal), followed by the Alexa Fluor 546 conjugated Donkey anti-Rabbit IgG (Invitrogen, Waltham, MA, USA). Cell nuclis were stained with DAPI. Images were collected on a Leica TCS SP8 confocal microscope.
2.5. Coimmunoprecipitation (co-IP) Assay
Co-IP assays were performed in HEK293T cells for their high transfection efficiency. The cells seeded in 6-well plates were cotransfected with 3Flag-tagged plasmid and 3HA-tagged plasmid (1.25 μg for each). An empty vector was used as a control. The cells were collected at 36 h post-transfection (hpt) and lysed with radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China) containing PMSF and protease inhibitor (MCE, Shanghai, China). The cell lysates were centrifuged and the supernatants were incubated with anti-Flag affinity gel (Sigma) for 4 h at 4 °C. The precipitates were washed with ice-cold PBST five times and subjected to Western blot as described above. Commercial anti-Flag (Sigma, Waltham, MA, USA) and anti-HA (CST, Danvers, MA, USA) antibodies were used.
2.6. RNA Interference
Three siRNAs targeting Mesocricetus auratus MSH2 (GenBank accession number: XM_005077304) were designed and synthesized (GenePharma, Shanghai, China), which were transfected into BHK-21 cells at a concentration of 25 nM using Lipofectamine RNAiMAX (Thermo Fisher, Waltham, MA, USA). A commercial synthesized NC siRNA was used as a negative control. The transfected cells were collected at 36 hpt to perform a Western blot analysis. Alternatively, the cells were infected with ADRV at an MOI of 0.5 and collected at the indicated time points to perform RNA or DNA extraction. The equences of the siRNAs are listed in Table 1.

Table 1.
siRNA sequences used in the present study.
2.7. MSH2 Knockout Cells Construction and Virus Infection
MSH2 knockout cells were established by the CRISPR-Cas9 method. Sequences targeting the Mesocricetus auratus MSH2 gene were designed and ligated into the plasmid pU6gRNA1Cas9EGFPU6gRNA2 (GenePharma, Shanghai, China) which contained two U6 promoters and can express two gRNAs, Cas9, and EGFP simultaneously. The two targeted sequences in the MSH2 gene were as follows: 5′-agaagtcgccgcggtcgaag-3′ (108–127, antisense) and 5′-cttctacacggcgcacggcg-3′ (123–142, sense). The two gRNAs would target the sequences from 108 to 142 bp of the MSH2 gene, which could enhance the editing efficiency and accuracy of one gRNA. The obtained plasmid was transfected into BHK-21 cells. At 48 hpt, the cells expressing EGFP were sorted into 96-well plates by flow cytometer and each well only contained one cell. The plates were cultured at 37 °C until clones formed. Individual clones were further transferred into 6-well plates and then identified by DNA sequencing and Western blotting. The finally obtained cell clone was named BHK-ΔMSH2.
For virus infection, wild-type BHK-21 or the BHK-ΔMSH2 were seeded into 24-well plates in the same cell numbers for each well. Then, the cells were inoculated with ADRV or ADRV46R-3Flag [14] at an MOI of 0.5. After incubation for 1 h, the medium containing the virus was moved and the fresh medium was added and incubated at 28 °C. At indicated time points, the cells were observed under a fluorescence microscope or collected for viral titer assay, or RNA and DNA extraction respectively.
2.8. Virus Titer Assay
Virus titers of the samples collected above were determined on triplicate monolayers of GSTC cells by using the 50% tissue culture infectious dose (TCID50) assays as described previously [25].
2.9. Real-Time RT-PCR for Virus Gene Expression
RNA was extracted from the collected cells with TRIzol Reagent (Thermo Fisher, Waltham, MA, USA). First-strand cDNA synthesis was performed using HiScript III RT SuperMix for qPCR (+gDNA wiper) (Vazyme, Nanjing, China) as per the manufacturer’s recommendations. RT-qPCR was conducted using a CFX96 Touch Real-Time PCR Detection System (BioRad, Hercules, CA, USA). Each RT-qPCR mixture contained 10 μL of SYBR Premix (2×), 0.5 μL of forward and reverse primers (for each primer), 1 μL of cDNA, and 8 μL of ultrapure water. The β-actin gene was used as an internal control. The RT-qPCR conditions were as follows: 95 °C for 10 min; 40 cycles of 95 °C for 15 s, 60 °C for 1 min; and a melt curve analysis at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s. The mRNA relative expression ratios of the treated group versus that of the control group were calculated by the 2−ΔΔCT method. The primers used in the previous study were used [14].
2.10. Real-Time PCR for Virus Genomic Copies
DNA was extracted with the Takara MiniBEST Universal Genomic DNA Extraction Kit (TakaRa, Maebashi, Japan). Virus genomic copies were determined by detecting the viral major capsid protein gene (MCP) copy numbers with quantitative real-time PCR (qPCR), which was conducted in a CFX96 Touch Real-Time PCR Detection System (BioRad) as described above. The plasmid pMD18T-MCP used previously was used as a template to construct the standard curve [14]. The MCP amount (genome copy number) was calculated with the standard curve.
2.11. Statistical Analysis
All data represent results of three biological replicates and were analyzed with Student’s t-test in GraphPad Prism 9 or Microsoft Excel. Significant differences are marked with a (0.01 < p < 0.05) or b (p < 0.01).
3. Results
3.1. MSH2 Was Expressed during ADRV Infection and Localized with Viral Nascent DNA
The expression of MSH2 during ADRV infection of BHK-21 cells was examined by Western blot analysis. The results showed that bands corresponding to MSH2 were stably detected among the examined times (0–48 hpi) (Figure 1A), which indicated that the expression of MSH2 was not inhibited by ADRV infection.
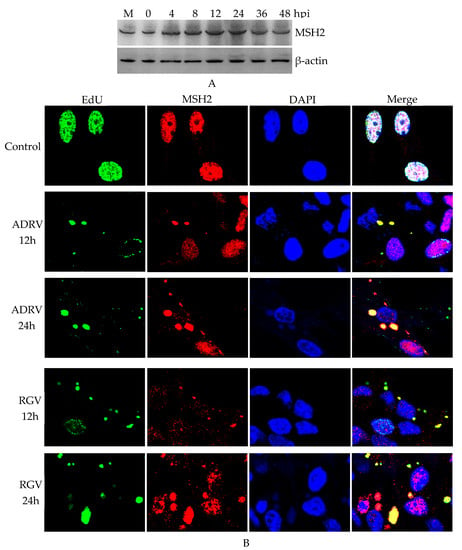
Figure 1.
Expression and subcellular localization of MutS homolog 2 (MSH2) in virus-infected BHK-21 cells. (A). Western blot analysis of MSH2 expression in Andrias davidianus ranavirus (ADRV) infected cells. (B). Immunofluorescence detection of MSH2 in ADRV and Rana grylio virus (RGV) infected cells. Nascent DNA was stained by EdU (green color). MSH2 was stained by rabbit anti-MSH2 antibody (Red color). DAPI (blue color) was used to stain the nuclei and viral factories. Objective magnification ×63.
Subcellular localization of MSH2 was further examined by immunofluorescence. EdU was used to label the virus nascent DNA and viral factories. As shown in Figure 1B, in the cells without infection (control), EdU (green color) was detected in the nucleus, indicating a replicating cell. MSH2 was also localized in the nucleus in the normal cells. However, in ADRV infected cells, most of EdU labeled loci were localized in the cytoplasm in an aggregated form, indicating the replicating viral DNA. MSH2 also appeared in the cytoplasm and colocalized with the cytoplasmic EdU and DAPI stained viral factories in the infected cells. Most of the fluorescence signal and size of the cytoplasmic colocalized EdU and MSH2 increased from 12 to 24 hpi. We also examined the subcellular localization of MSH2 in BHK-21 cells infected by another ranavirus RGV, which showed similar results as the ADRV infected cells (Figure 1B). Collectively, expression and colocalization assays indicated that host MSH2 could participate in ADRV infection.
3.2. MSH2 Interacted with Viral Proteins
The MSH2 localized in the cytoplasmic viral factories in infected cells while its normal localization was the nucleus, which indicated possible interactions between MSH2 and viral proteins. We further tried to explore the interacted viral proteins. With previous transcriptome data, the MSH2 coding sequence was first cloned from GSTC cells. The ORF of MSH2 of GSTC (gMSH2) has a size of 2796 bp (GenBank accession number ON086763), encoding a protein with 931 aa.
Using the NanoLuciferase complementation assay established previously [14], a relatively higher luciferase was found when cotransfection of gMSH2 with ADRV 35L, 47L, or 98R (data not shown), indicating possible interactions among them. Co-IP was further performed to verify the interactions. As shown in Figure 2, the positive bands corresponding to 3HA-MSH2 were detected in immunoprecipitated (IP) complex (affinity by anti-Flag gel) from the samples co-transfected with 35L-3Flag, 47L-3Flag, and 98R-3Flag respectively. In the three combinations, the IP band of 3HA-MSH2 was strongest in 35L-3Flag cotransfected cells and weakest in 98R cotransfected cells.
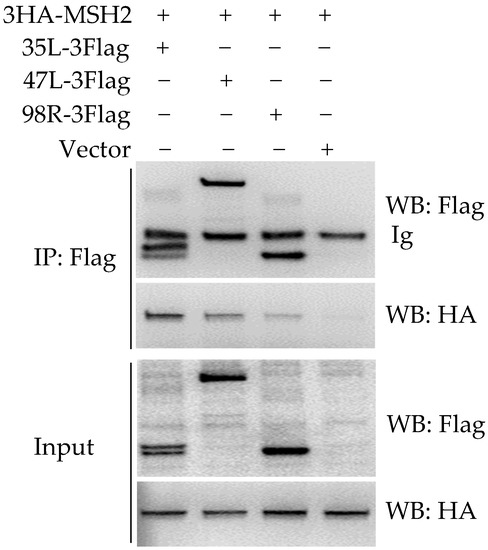
Figure 2.
Detection of the interaction between MSH2 and ADRV-35L, ADRV-47L, or ADRV-98R. Cell lysates (Input) from HEK293T cells cotransfected with plasmids expressing the indicated proteins and immunoprecipitated (IP) protein complexes were subjected to Western blot analysis with anti-Flag or anti-HA antibody.
3.3. Knockdown of MSH2 Affected Viral Late Gene Expression
We further performed an RNAi assay to test the effect of MSH2 knockdown on virus infection. In the three siRNAs targeted to MSH2, siMSH2-1 and siMSH2-2 showed a relatively higher interference efficiency than siMSH2-3 as revealed by Western blot analysis (Figure 3A). siMSH2-1 and siMSH2-2 were used in the following analysis. RT-qPCR showed that there was no significant difference in the expression of viral immediate-early gene ICP18 between the two siRNAs and control NC at 12 and 24 hpi (Figure 3B). However, the two siRNAs both significantly (p = 0.010401 and 0.000128 for siMSH2-1, p = 0.000414 and 0.000109 for siMSH2-2, at 12 and 24 hpi respectively) reduced the expression of the viral late gene MCP (Figure 3C). The viral genome copy numbers were further detected by quantitative qPCR. The results showed that there was no significant difference between the siRNAs and NC at 6 hpi, and the viral genome copy numbers in siRNA transfected cells were less than that of NC at 12 and 24 hpi, but the significant difference was only observed in siRNA-2 transfected cells at 24 hpi (p = 0.017361) (Figure 3D).
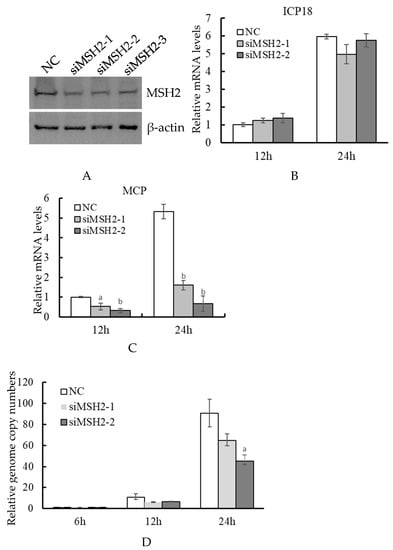
Figure 3.
Determination of the effect of MSH2 knockdown on ADRV infection. siRNAs targeting MSH2 were transfected into BHK-21 cells. The transfected cells were subjected to Western blotting analysis (A) or infected with ADRV. Expression of ADRV immediate early gene ICP18 (B) and late gene MCP (C) were determined by RT-qPCR. The virus genome copy numbers were determined by quantitative real-time PCR with the standard curve. The virus genome copy number in NC transfected cells at 6 hpi was set as 1 in the figure (D). Significant differences (versus NC at the time point) are marked with a (0.01 < p < 0.05) or b (p < 0.01).
3.4. Construction of MSH2 Knockout Cells
Considering the efficiency of the MSH2 RNAi was not high enough, there were still a large amount of expressed MSH2, which could affect the RNAi results, therefore, we tried to establish MSH2 knockout cells by CRISPR/Cas9. A BHK-21 cell lacking the expression of MSH2 (BHK-ΔMSH2) was finally obtained. The sgRNAs targeted sequence was located from 108 to 142 nt in the MSH2 mRNA. DNA sequencing showed that the N terminal region of the MSH2 coding sequence was mutated (Figure 4A). The MSH2 corresponding band was not detected in the BHK-ΔMSH2 cells (Figure 4B).
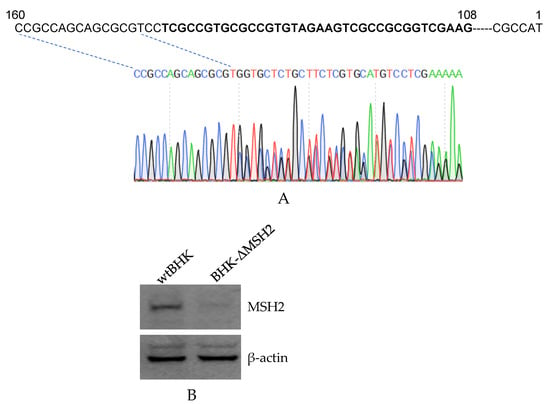
Figure 4.
Establishment of MSH2 knockout BHK cells by CRISPR/Cas9. (A). DNA sequencing confirmation of the mutated regions in the MSH2 gene. The sequences with bold font are the targeted sequences by the two gRNAs. The PCR products were sequenced from the reverse direction and the sequence chromatogram showed the sequence from position 160 bp of the gene. (B). Western blot analysis of MSH2 expression in wild-type BHK-21 (wtBHK) and mutated BHK (BHK-ΔMSH2) cells. Detection of β-actin was used as an internal control.
3.5. MSH2 Knockout Inhibited ADRV Infection
ADRV infection in the BHK-ΔMSH2 cells was examined by infecting the cells with a recombinant ADRV expression EGFP. The results showed that EGFP expression was reduced in BHK-ΔMSH2 cells compared to wild-type BHK cells at the examined time points (24 and 48 hpi) (Figure 5A). The virus titers at different times (0, 12, 24, 48 hpi) were further determined by the TCID50 assay. The virus titers between wild-type BHK and BHK-ΔMSH2 at 0 hpi were nearly equivalent, but the virus titers from BHK-ΔMSH2 were significantly less than that from wild-type BHK at 12 (p = 0.000413), 24 (p = 0.005739), and 48 (p = 0.015251) hpi (Figure 5B), which indicated that MSH2 knockout inhibited ADRV infection.
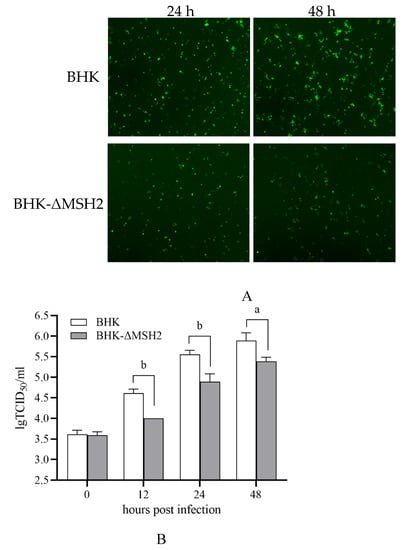
Figure 5.
MSH2 knockout inhibited ADRV infection. (A). Fluorescence observation of the wild type BHK and BHK-ΔMSH2 cells infected with ADRV expression EGFP at 24 and 48 hpi. Objective magnification ×4. (B). Determination of virus titers in the two cells infected with ADRV by TCID50 methods showed that ADRV titers were reduced in MSH2 knockout cells. Significant differences are marked with a (0.01 < p < 0.05) or b (p < 0.01).
3.6. MSH2 Knockout Reduced Viral Genome Replication and Late Gene Expression
The virus gene expression and genome replication were further examined in the BHK-ΔMSH2 cells. Similar to that observed in RNAi assays, the expression of ADRV immediate early gene ICP18 was not affected in BHK-ΔMSH2 cells compared to wild-type BHK cells at the tested time points (2, 4, 6, and 12 hpi) (Figure 6A), and the expression of the late gene MCP was significantly reduced at 12 (p = 0.008371) and 24 hpi (p = 0.000757) in the BHK-ΔMSH2 cells (Figure 6B).
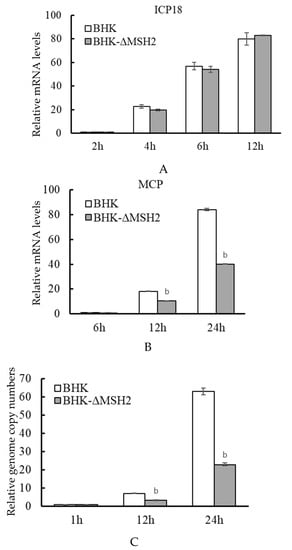
Figure 6.
MSH2 knockout inhibited genome replication and late gene expression of ADRV. Expression of the immediate-early gene ICP18 (A) and late gene MCP (B), and the virus genome copy numbers (C) were determined as described above. Significant differences (ΔMSH2 versus BHK-1 at the time point) are marked with b (p < 0.01).
The virus genome copy numbers were determined simultaneously. No significant difference was found between the BHK-ΔMSH2 and wild-type BHK cells at 1 hpi, which indicated that MSH2 knockout had no effect on virus attachment and entry. However, the viral genome copy numbers from BHK-ΔMSH2 cells were significantly reduced at 12 (p = 0.00397) and 24 hpi (p = 0.000271) compared to the wild type BHK cells (Figure 6C). These results indicated that MSH2 knockout affected ADRV genome replication and late gene expression.
4. Discussion
Host DNA repair protein MSH2 was detected in our previous study on ranavirus replisome and transcription complex [14], although genome sequence analysis has revealed that iridoviruses including ranaviruses encoded protein potentially involved in DNA repair, such as the RAD2 homolog [6,26,27,28,29,30,31]. In the present study, the host MSH2 was cloned and characterized as an important factor for ADRV replication, which was thefirst to report the host mismatch repair protein involved in iridovirus infection.
Subcellular localization revealed that MSH2 localized in cytoplasmic viral factories is the place where viral DNA replication occurred [32]. Most of the viral nascent DNA colocalized with MSH2, which indicated the association between MSH2 and virus DNA replication. However, sometimes, there were still some punctate areas emitting weak green color (nascent DNA) and a strong red color (MSH2), which hinted that the nascent DNA synthesis and MSH2 localization might be asynchronous steps.
MSH2 was further shown to have interactions with ADRV proteins 35L, 47L, and 98R. The function of ADRV-98R was unknown up to now. In our previous study, ADRV-35L was found to be associated with the virus transcription complex [14], and ADRV-47L is the virus DNA polymerase [6,14]. The interactions hinted that MSH2 might participate in virus genome replication and gene transcription, but the exact mechanism needs further research.
RNAi assay revealed that knockdown of MSH2 inhibited the expression of the viral late gene but not the immediate-early gene, which indicated that MSH2 participated in a later step in the virus life cycle. It has been reported that silencing of MSH2 inhibited early gene expression in HSV infected cells [20], which is a virus replicating in the cell nucleus. The effect on virus DNA replication was not obvious in the present RNAi assay, which prompted us to deplete the MSH2 expression, and finally, the BHK-21 cells that lacked MSH2 expression were obtained. Deletion of MSH2 expression significantly affected the virus’ late gene expression and DNA replication. However, it has been reported that full late gene expression requires prior DNA synthesis (nascent DNA) [1]. So, the transcription of late genes of ranaviruses started after virus DNA replication [33], and inhibition of virus DNA replication would affect the transcription of late genes. Thus, it is not clear whether MSH2 participated in virus late gene transcription directly.
In addition, BHK-21 cells have been used in the research of ranavirus infection, such as frog virus 3 [34,35]. Our previous experiments have shown that ADRV and RGV infected efficiently in BHK-21 cells. The cells have been used in the investigation of the transcription complex of ADRV [14]. So, we performed the subcellular localization, knockdown, and knockout assays of MSH2 in BHK cells for the availability of commercial antibodies and the feasibility of gene knockout by CRISPR/Cas9, although the MSH2 was initially found in ADRV infected GSTC cells as the viral nascent DNA associated protein. The results from BHK-21 cells also suggested the importance of MSH2 in ADRV replication in different cells.
Therefore, the present study confirmed that cellular MSH2 was hijacked by ADRV to facilitate its efficient replication and transcription by using subcellular localization, co-IP, and gene knockdown/knockout assays, which could occur in other ranavirus infection for the subcellular co-localization was also observed in RGV infection. Thus, the study provided new insights into the understanding of ranavirus replication and ranavirus-host interactions.
Author Contributions
Conceptualization, F.K. and Q.Z.; methodology, F.K., R.W. and Z.W.; writing—original draft preparation, F.K.; writing—review and editing, F.K. and Q.Z.; funding acquisition, F.K. and Q.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (31972839), the National Key R&D Plan of the Ministry of Science and Technology, China (2018YFD0900302), the Strategic Pilot Science and Technology of the Chinese Academy of Sciences Project (XDA24030203), the Project from Chinese Academy of Sciences (KJZD-SW-L11), and the Institute of Hydrobiology (Y85Z02-1-3-1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The MSH2 sequence of GSTC cells obtained in the study has been submitted into GenBank under accession number ON086763.
Acknowledgments
We would like to thank Mo-Han Cao for his technical assistance, and Fang Zhou and Yan Wang at The Analysis and Testing Center of Institute of Hydrobiology, Chinese Academy of Sciences for assistances with confocal microscopy analysis and flow cytometer cell sorting.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Chinchar, V.G.; Hick, P.; Ince, I.A.; Jancovich, J.K.; Marschang, R.; Qin, Q.; Subramaniam, K.; Waltzek, T.B.; Whittington, R.; Williams, T.; et al. ICTV virus taxonomy profile: Iridoviridae. J. Gen. Virol. 2017, 98, 890–891. [Google Scholar] [CrossRef]
- Chinchar, V.G.; Waltzek, T.B.; Subramaniam, K. Ranaviruses and other members of the family Iridoviridae: Their place in the virosphere. Virology 2017, 511, 259–271. [Google Scholar] [CrossRef]
- Bandin, I.; Dopazo, C.P. Host range, host specificity and hypothesized host shift events among viruses of lower vertebrates. Vet. Res. 2011, 42, 67. [Google Scholar] [CrossRef] [Green Version]
- Carstairs, S.J.; Kyle, C.J.; Vilaca, S.T. High prevalence of subclinical frog virus 3 infection in freshwater turtles of Ontario, Canada. Virology 2020, 543, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Y.; Zhao, Z.; Xiao, F.; Li, Z.-Q.; Gui, J.-F. Molecular characterization of three Rana grylio virus (RGV) isolates and Paralichthys olivaceus lymphocystis disease virus (LCDV-C) in iridoviruses. Aquaculture 2006, 251, 1–10. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Gui, J.F.; Gao, X.C.; Pei, C.; Hong, Y.J.; Zhang, Q.Y. Genome architecture changes and major gene variations of Andrias davidianus ranavirus (ADRV). Vet. Res. 2013, 44, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.Y.; Gui, J.F. Virus genomes and virus-host interactions in aquaculture animals. Sci. China Life Sci. 2015, 58, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Gui, L.; Chinchar, V.G.; Zhang, Q.Y. Molecular basis of pathogenesis of emerging viruses infecting aquatic animals. Aquac. Fish. 2018, 3, 1–5. [Google Scholar] [CrossRef]
- Chinchar, V.G.; Waltzek, T.B. Ranaviruses: Not just for frogs. PLoS Path. 2014, 10, e1003850. [Google Scholar] [CrossRef] [Green Version]
- Goorha, R. Frog Virus 3 DNA Replication Occurs in Two Stages. J. Virol. 1982, 43, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Jancovich, J.K.; Qin, Q.W.; Zhang, Q.Y.; Chinchar, V.G. Ranavirus replication: Molecular, cellular, and immunological events. In Ranaviruses Lethal Pathogens of Ectothermic Vertebrates; Gray, M.J., Chinchar, V.G., Eds.; Springer: New York, NY, USA, 2015; pp. 105–139. [Google Scholar]
- Zeng, X.T.; Zhang, Q.Y. Interaction between two iridovirus core proteins and their effects on ranavirus (RGV) replication in cells from different species. Viruses 2019, 11, 416. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Ke, F.; Gui, J.; Zhang, Q. Characterization of an early gene encoding for dUTPase in Rana grylio virus. Virus Res. 2007, 123, 128–137. [Google Scholar] [CrossRef]
- Ke, F.; Yu, X.-D.; Wang, Z.-H.; Gui, J.-F.; Zhang, Q.-Y. Replication and transcription machinery for ranaviruses: Components, correlation, and functional architecture. Cell Biosci. 2022, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Nei, M.; Ma, H. The origins and early evolution of DNA mismatch repair genes--multiple horizontal gene transfers and co-evolution. Nucleic Acids Res. 2007, 35, 7591–7603. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. Rev. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daikoku, T.; Kudoh, A.; Sugaya, Y.; Iwahori, S.; Shirata, N.; Isomura, H.; Tsurumi, T. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. J. Biol. Chem. 2006, 281, 11422–11430. [Google Scholar] [CrossRef] [Green Version]
- Mohni, K.N.; Mastrocola, A.S.; Bai, P.; Weller, S.K.; Heinen, C.D. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J. Virol. 2011, 85, 12241–12253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Xiao, F.; Li, Z.Q.; Gui, J.F.; Mao, J.; Chinchar, V.G. Characterization of an iridovirus from the cultured pig frog Rana grylio with lethal syndrome. Dis. Aquat. Organ. 2001, 48, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.D.; Chen, Z.Y.; Huang, X.; Gao, X.C.; Zhang, Q.Y. Establishment of three cell lines from Chinese giant salamander and their sensitivities to the wild-type and recombinant ranavirus. Vet. Res. 2015, 46, 58. [Google Scholar] [CrossRef] [Green Version]
- Ke, F.; Gui, J.F.; Chen, Z.Y.; Li, T.; Lei, C.K.; Wang, Z.H.; Zhang, Q.Y. Divergent transcriptomic responses underlying the ranaviruses-amphibian interaction processes on interspecies infection of Chinese giant salamander. BMC Genom. 2018, 19, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, F.; Wang, Z.H.; Ming, C.Y.; Zhang, Q.Y. Ranaviruses bind cells from different species through interaction with heparan sulfate. Viruses 2019, 11, 593. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.T.; Gao, X.C.; Zhang, Q.Y. Rana grylio virus 43R encodes an envelope protein involved in virus entry. Virus Genes 2018, 54, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.G.; Barkman, T.J.; Gregory Chinchar, V.; Essani, K. Comparative genomic analyses of frog virus 3, type species of the genus Ranavirus (family Iridoviridae). Virology 2004, 323, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.Y.; Ou, T.; Zhu, R.L.; Zhang, Q.Y. Sequencing and analysis of the complete genome of Rana grylio virus (RGV). Arch. Virol. 2012, 157, 1559–1564. [Google Scholar] [CrossRef]
- Song, W.J.; Qin, Q.W.; Qiu, J.; Huang, C.H.; Wang, F.; Hew, C.L. Functional genomics analysis of Singapore grouper iridovirus: Complete sequence determination and proteomic analysis. J. Virol. 2004, 78, 12576–12590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Chen, M.M.; Wang, R.Y.; Wan, X.Y.; Li, C.; Zhang, Q.L.; Dong, X.; Yang, B.; Xiang, J.H.; Huang, J. Complete genome sequence of shrimp hemocyte iridescent virus (SHIV) isolated from white leg shrimp, Litopenaeus vannamei. Arch. Virol. 2018, 163, 781–785. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Xiao, F.; Xie, J.; Li, Z.Q.; Gui, J.F. Complete genome sequence of lymphocystis disease virus isolated from China. J. Virol. 2004, 78, 6982–6994. [Google Scholar] [CrossRef] [Green Version]
- He, J.G.; Deng, M.; Weng, S.P.; Li, Z.; Zhou, S.Y.; Long, Q.X.; Wang, X.Z.; Chan, S.M. Complete genome analysis of the mandarin fish infectious spleen and kidney necrosis iridovirus. Virology 2001, 291, 126–139. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.H.; Huang, Y.H.; Yuan, X.P.; Zhang, Q.Y. Electron microscopic examination of the viromatrix of Rana grylio virus in a fish cell line. J. Virol. Methods 2006, 133, 117–123. [Google Scholar] [CrossRef]
- Majji, S.; Thodima, V.; Sample, R.; Whitley, D.; Deng, Y.; Mao, J.; Chinchar, V.G. Transcriptome analysis of Frog virus 3, the type species of the genus Ranavirus, family Iridoviridae. Virology 2009, 391, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.M.; Bravo, R.; Kelly, D.C. Frog virus 3 replication: Analysis of structural and nonstructural polypeptides in infected BHK cells by acidic and basic two-dimensional gel electrophoresis. J. Virol. 1980, 33, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunwald, J.; Nonnenmacher, H.; Tripier-Darcy, F. Ultrastructural and biochemical study of frog virus 3 uptake by BHK-21 cells. J. Gen. Virol. 1985, 66 Pt 2, 283–293. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).