
Chirohepevirus from Bats: Insights into Hepatitis E Virus Diversity and Evolution


Abstract
:1. Introduction
2. HEV-D and Bats
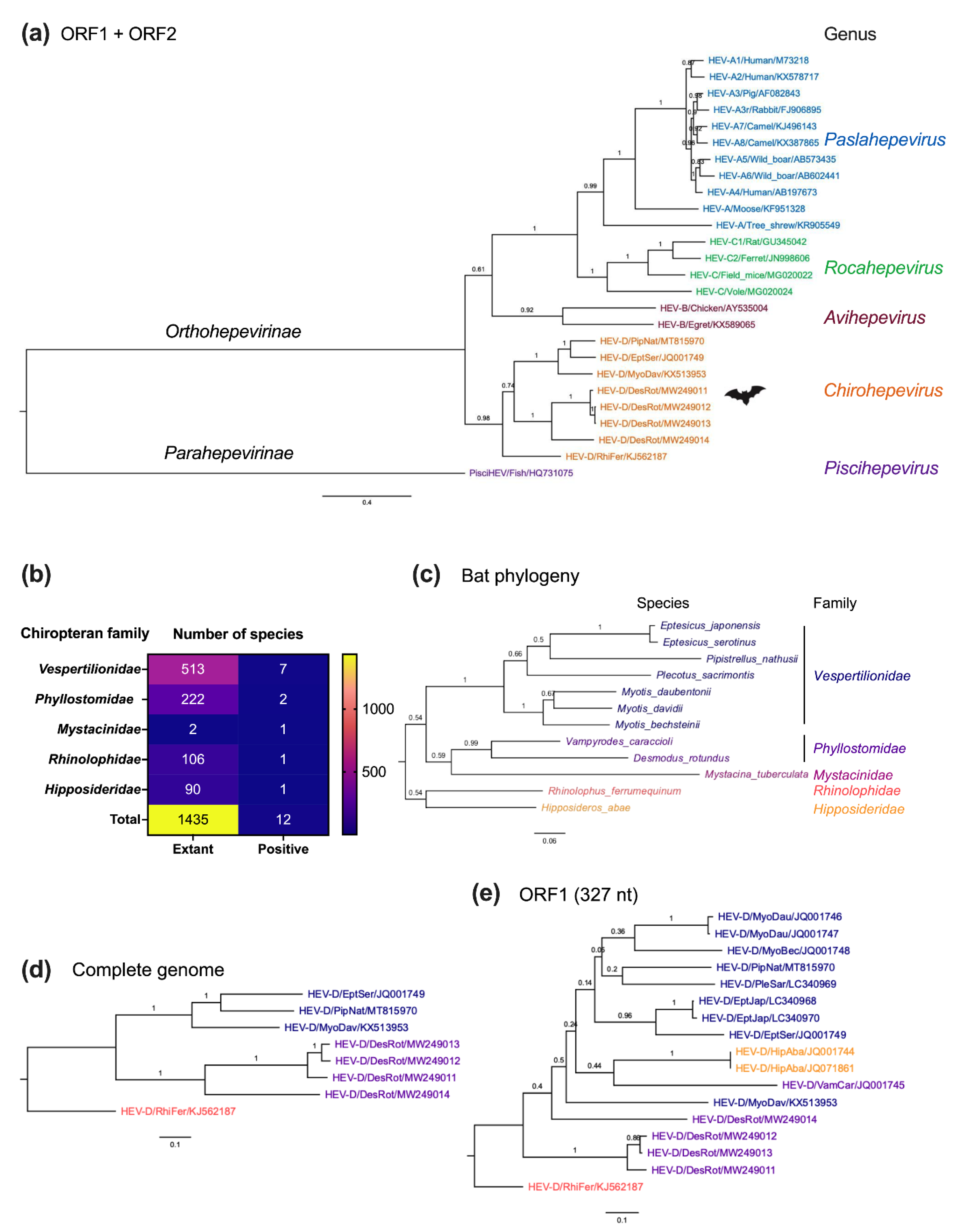
3. Genetic Diversity of Bat HEV-D
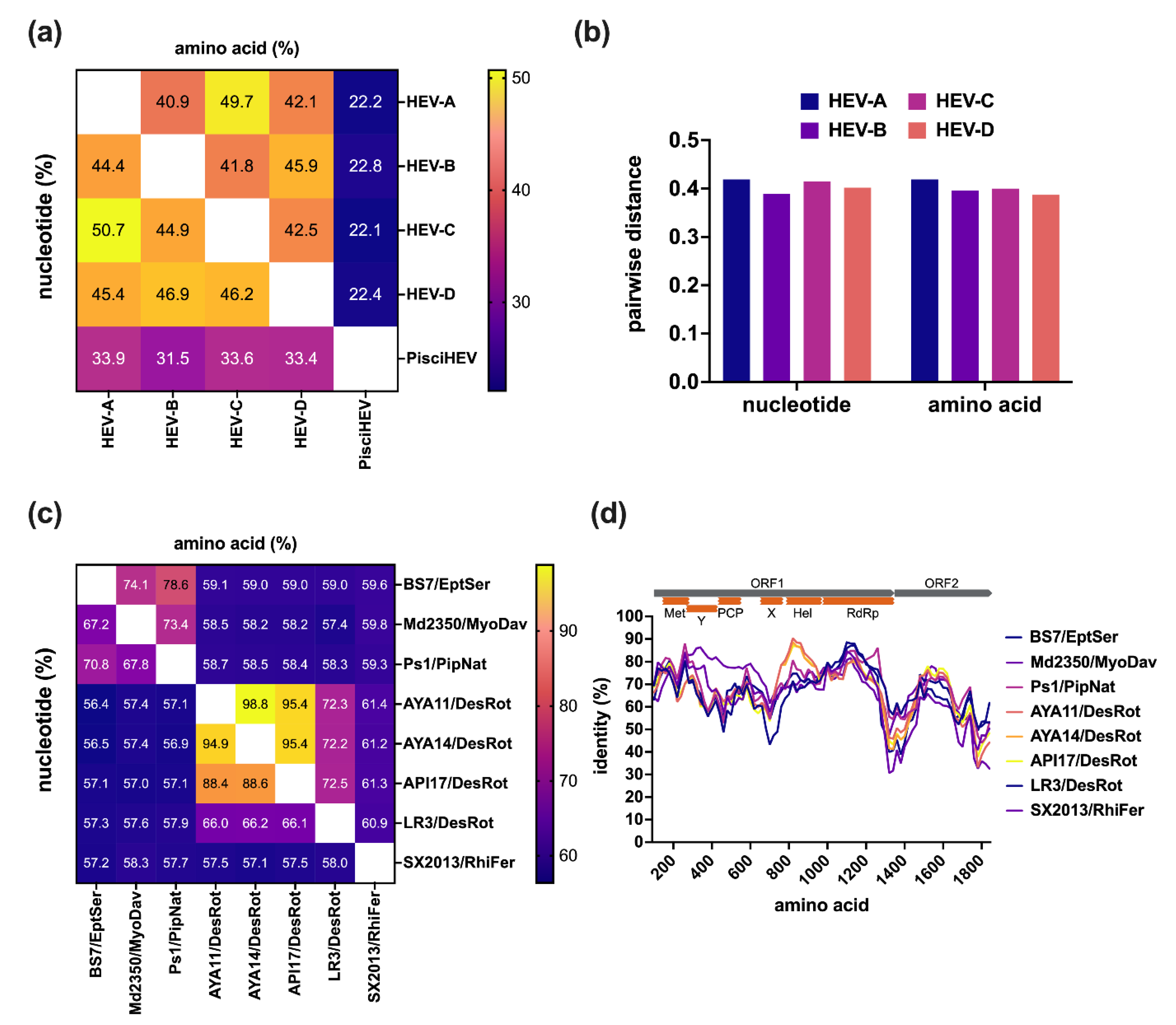
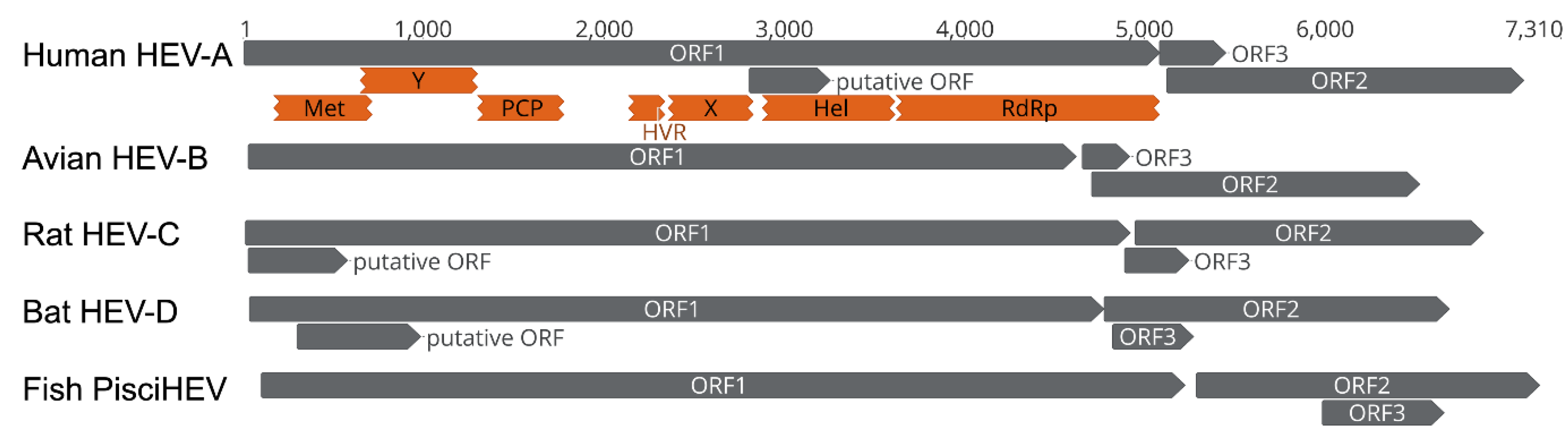
4. Genomic Characterization of Bat HEV-D
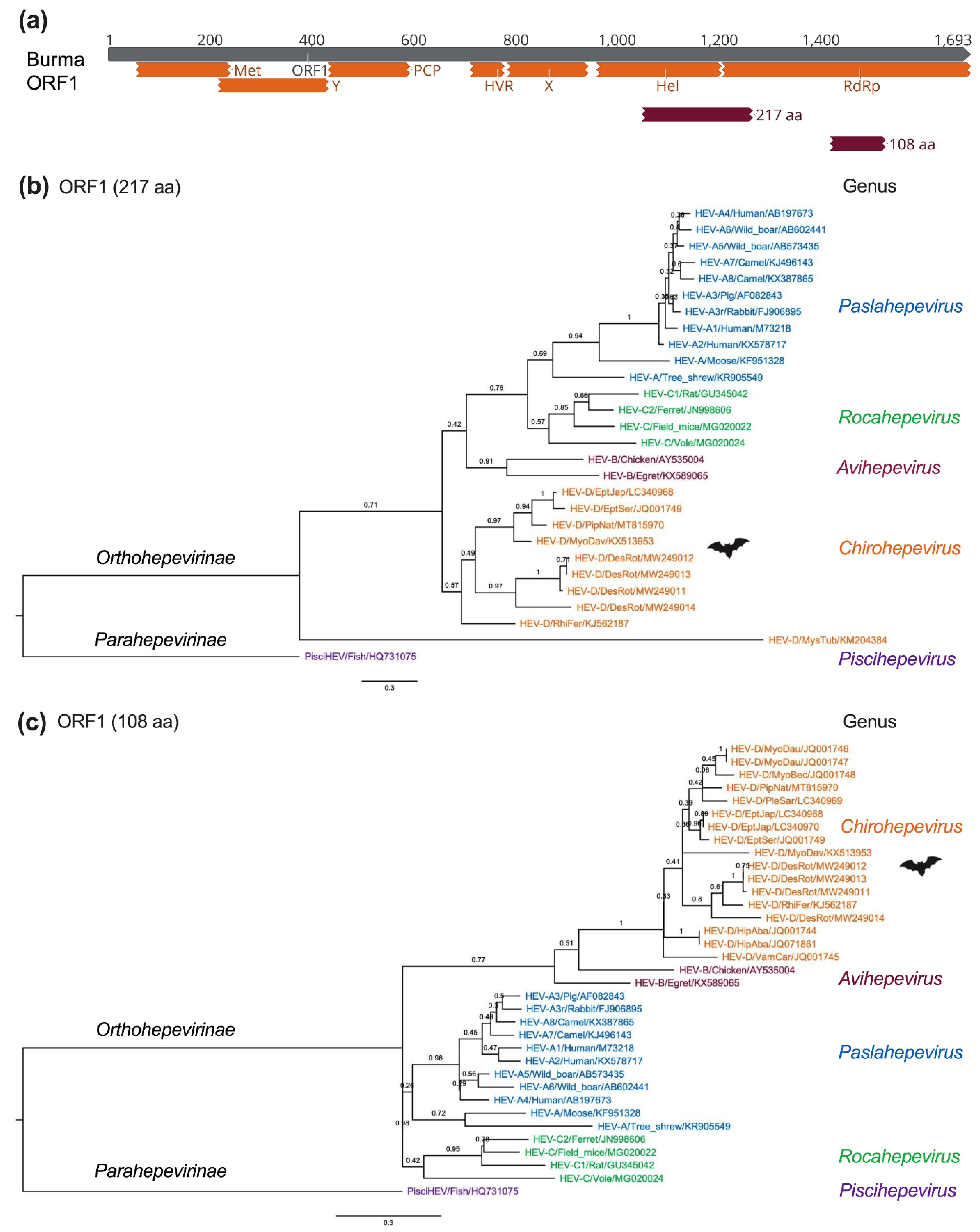
5. Molecular Evolution of Bat HEV-D
6. Infection Patterns of Bat HEV-D
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO. Viral Hepatitis in the WHO South-East Asia Region. 2011. Available online: https://apps.who.int/iris/bitstream/handle/10665/206521/B4752.pdf?sequence=1&isAllowed=y (accessed on 8 February 2022).
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, R.H.; Dusheiko, G.; Williamson, C. Pregnancy and liver disease. J. Hepatol. 2016, 64, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuroo, M.S. Hepatitis E and Pregnancy: An Unholy Alliance Unmasked from Kashmir, India. Viruses 2021, 13, 1329. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J. Hepatitis E Virus: An Emerging Zoonotic Virus Causing Acute and Chronic Liver Disease. In Liver: Biology and Pathobiology, 6th ed.; Wiley: Hoboken, NJ, USA, 2020; pp. 915–925. [Google Scholar]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Mechanism of Cross-Species Transmission, Adaptive Evolution and Pathogenesis of Hepatitis E Virus. Viruses 2021, 13, 909. [Google Scholar] [CrossRef]
- Todt, D.; Meister, T.L.; Steinmann, E. Hepatitis E virus treatment and ribavirin therapy: Viral mechanisms of nonresponse. Curr. Opin. Virol. 2018, 32, 80–87. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.-J. Therapeutic targets for the treatment of Hepatitis E virus infection. Expert Opin. Ther. Targets 2015, 19, 1245–1260. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Marion, O.; Abravanel, F.; Izopet, J.; Dalton, H.R. Extrahepatic manifestations of Hepatitis E virus. Liver Int. 2016, 36, 467–472. [Google Scholar] [CrossRef]
- Lhomme, S.; Abravanel, F.; Cintas, P.; Izopet, J. Hepatitis E Virus Infection: Neurological Manifestations and Pathophysiology. Pathogens 2021, 10, 1582. [Google Scholar] [CrossRef]
- Khuroo, M.S. Study of an epidemic of non-A, non-B hepatitis: Possibility of another human hepatitis virus distinct from post-transfusion non-A, non-B type. Am. J. Med. 1980, 68, 818–824. [Google Scholar] [CrossRef]
- Balayan, M.S.; Andjaparidze, A.G.; Savinskaya, S.S.; Ketiladze, E.S.; Braginsky, D.M.; Suavinov, A.P.; Poleschuk, V.F. Evidence for a Virus in Non-A, Non-B Hepatitis Transmitted via the Fecal-Oral Route. Intervirology 1983, 20, 23–31. [Google Scholar] [CrossRef]
- Reyes, G.R.; Purdy, M.A.; Kim, J.P.; Luk, K.-C.; Young, L.M.; Fry, K.E.; Bradley, D.W. Isolation of a cDNA from the Virus Responsible for Enterically Transmitted Non-A, Non-B Hepatitis. Science 1990, 247, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J.; Purcell, R.H.; Halbur, P.G.; Lehman, J.R.; Webb, D.M.; Tsareva, T.S.; Haynes, J.S.; Thacker, B.J.; Emerson, S.U. A novel virus in swine is closely related to the human Hepatitis E virus. Proc. Natl. Acad. Sci. USA 1997, 94, 9860–9865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.-J.; Halbur, P.G.; Shapiro, M.S.; Govindarajan, S.; Bruna, J.D.; Mushahwar, I.K.; Purcell, R.H.; Emerson, S.U. Genetic and Experimental Evidence for Cross-Species Infection by Swine Hepatitis E Virus. J. Virol. 1998, 72, 9714–9721. [Google Scholar] [CrossRef] [Green Version]
- Doceul, V.; Bagdassarian, E.; Demange, A.; Pavio, N. Zoonotic Hepatitis E Virus: Classification, Animal Reservoirs and Transmission Routes. Viruses 2016, 8, 270. [Google Scholar] [CrossRef]
- Izopet, J.; Tremeaux, P.; Marion, O.; Migueres, M.; Capelli, N.; Chapuy-Regaud, S.; Mansuy, J.-M.; Abravanel, F.; Kamar, N.; Lhomme, S. Hepatitis E virus infections in Europe. J. Clin. Virol. 2019, 120, 20–26. [Google Scholar] [CrossRef]
- Wang, B.; Harms, D.; Papp, C.P.; Niendorf, S.; Jacobsen, S.; Lütgehetmann, M.; Pischke, S.; Wedermeyer, H.; Hofmann, J.; Bock, C.-T. Comprehensive Molecular Approach for Characterization of Hepatitis E Virus Genotype 3 Variants. J. Clin. Microbiol. 2018, 56, e01686-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Hofmann, J.; Köhler, A.; Wang, B.; Bock, C.-T.; Schott, E.; Reinke, P.; Nickel, P. Prevalence and Clinical Correlates of Chronic Hepatitis E Infection in German Renal Transplant Recipients With Elevated Liver Enzymes. Transplant. Direct 2018, 4, e341. [Google Scholar] [CrossRef]
- Kamar, N.; Selves, J.; Mansuy, J.-M.; Ouezzani, L.; Péron, J.-M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E Virus and Chronic Hepatitis in Organ-Transplant Recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Grange, Z.L.; Goldstein, T.; Johnson, C.K.; Anthony, S.; Gilardi, K.; Daszak, P.; Olival, K.J.; O’Rourke, T.; Murray, S.; Olson, S.H.; et al. Ranking the risk of animal-to-human spillover for newly discovered viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2002324118. [Google Scholar] [CrossRef]
- Wang, B.; Meng, X.-J. Hepatitis E virus: Host tropism and zoonotic infection. Curr. Opin. Microbiol. 2021, 59, 8–15. [Google Scholar] [CrossRef]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.-J.; Okamoto, H.; Van Der Poel, W.H.M.; Smith, D.B. Ictv Report Consortium, ICTV Virus Taxonomy Profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.P. The Current Host Range of Hepatitis E Viruses. Viruses 2019, 11, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Genetic Variability and Evolution of Hepatitis E Virus. Viruses 2019, 11, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Harms, D.; Yang, X.-L.; Bock, C.-T. Orthohepevirus C: An Expanding Species of Emerging Hepatitis E Virus Variants. Pathogens 2020, 9, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, G.; Boros, A.; Pankovics, P. Review of Hepatitis E Virus in Rats: Evident Risk of Species Orthohepevirus C to Human Zoonotic Infection and Disease. Viruses 2020, 12, 1148. [Google Scholar] [CrossRef]
- Rasche, A.; Sander, A.-L.; Corman, V.M.; Drexler, J.F. Evolutionary biology of human hepatitis viruses. J. Hepatol. 2019, 70, 501–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Meng, X.-J. Structural and molecular biology of hepatitis E virus. Comput. Struct. Biotechnol. J. 2021, 19, 1907–1916. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.-J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9, a031724. [Google Scholar] [CrossRef]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [Green Version]
- LeDesma, R.; Nimgaonkar, I.; Ploss, A. Hepatitis E Virus Replication. Viruses 2019, 11, 719. [Google Scholar] [CrossRef] [Green Version]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.-C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Drexler, J.F.; Seelen, A.; Corman, V.M.; Tateno, A.F.; Cottontail, V.; Zerbinati, R.M.; Gloza-Rausch, F.; Klose, S.M.; Adu-Sarkodie, Y.; Oppong, S.K.; et al. Bats Worldwide Carry Hepatitis E Virus-Related Viruses That Form a Putative Novel Genus within the Family Hepeviridae. J. Virol. 2012, 86, 9134–9147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velavan, T.P.; Pallerla, S.R.; Johne, R.; Todt, D.; Steinmann, E.; Schemmerer, M.; Wenzel, J.J.; Hofmann, J.; Shih, J.W.K.; Wedemeyer, H.; et al. Hepatitis E: An update on One Health and clinical medicine. Liver Int. 2021, 41, 1462–1473. [Google Scholar] [CrossRef]
- Wang, B.; Akanbi, O.A.; Harms, D.; Adesina, O.; Osundare, F.A.; Naidoo, D.; Deveaux, I.; Ogundiran, O.; Ugochukwu, U.; Mba, N.; et al. A new Hepatitis E virus genotype 2 strain identified from an outbreak in Nigeria, 2017. Virol. J. 2018, 15, 163. [Google Scholar] [CrossRef] [Green Version]
- Hoan, N.X.; Huy, P.X.; Sy, B.T.; Meyer, C.G.; Van Son, T.; Binh, M.T.; Giang, D.P.; Anh, D.T.; Bock, C.-T.; Wang, B.; et al. High Hepatitis E virus (HEV) Positivity Among Domestic Pigs and Risk of HEV Infection of Individuals Occupationally Exposed to Pigs and Pork Meat in Hanoi, Vietnam. Open Forum Infect. Dis. 2019, 6, ofz306. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.-H.; Tan, B.-H.; Teo, E.C.-Y.; Lim, S.-G.; Dan, Y.-Y.; Wee, A.; Aw, P.P.K.; Zhu, Y.; Hibberd, M.L.; Tan, C.-K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357.e353. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Simmonds, P. Classification and Genomic Diversity of Enterically Transmitted Hepatitis Viruses. Cold Spring Harb. Perspect. Med. 2018, 8, a031880. [Google Scholar] [CrossRef]
- Meng, X.-J. Expanding Host Range and Cross-Species Infection of Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005695. [Google Scholar] [CrossRef]
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many species of mammals are there? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, G.S.; Munshi-South, J. Life history, ecology and longevity in bats. Aging Cell 2002, 1, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Walker, P.J.; Poon, L.L. Mass extinctions, biodiversity and mitochondrial function: Are bats ‘special’ as reservoirs for emerging viruses? Curr. Opin. Virol. 2011, 1, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Wibbelt, G.; Moore, M.S.; Schountz, T.; Voigt, C.C. Emerging diseases in Chiroptera: Why bats? Biol. Lett. 2010, 6, 438–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.-F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Genet. 2006, 4, 23–35. [Google Scholar] [CrossRef]
- Yang, X.-L.; Hu, B.; Wang, B.; Wang, M.-N.; Zhang, Q.; Zhang, W.; Wu, L.-J.; Ge, X.-Y.; Zhang, Y.-Z.; Daszak, P.; et al. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2016, 90, 3253–3256. [Google Scholar] [CrossRef] [Green Version]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.-P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chretien, D.; Sanamxay, D.; et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef]
- Wang, L.-F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Tan, C.W.; Yang, X.; Anderson, D.E.; Wang, L.-F. Bat virome research: The past, the present and the future. Curr. Opin. Virol. 2021, 49, 68–80. [Google Scholar] [CrossRef]
- Pavlovich, S.S.; Lovett, S.P.; Koroleva, G.; Guito, J.C.; Arnold, C.; Nagle, E.R.; Kulcsar, K.; Lee, A.; Thibaud-Nissen, F.; Hume, A.; et al. The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell 2018, 173, 1098–1110.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yang, X.-L.; Li, W.; Zhu, Y.; Ge, X.-Y.; Zhang, L.-B.; Zhang, Y.-Z.; Bock, C.-T.; Shi, Z.-L. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol. J. 2017, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Murakami, S.; Yamamoto, T.; Mineshita, K.; Sakuyama, M.; Sasaki, R.; Maeda, K.; Horimoto, T. Detection of bat hepatitis E virus RNA in microbats in Japan. Virus Genes 2018, 54, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Moore, N.E.; Murray, Z.L.; McInnes, K.; White, D.J.; Tompkins, D.M.; Hall, R.J. Discovery of novel virus sequences in an isolated and threatened bat species, the New Zealand lesser short-tailed bat (Mystacina tuberculata). J. Gen. Virol. 2015, 96, 2442–2452. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Bergner, L.M.; Mollentze, N.; Orton, R.J.; Tello, C.; Broos, A.; Biek, R.; Streicker, D.G. Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats. Viruses 2021, 13, 252. [Google Scholar] [CrossRef]
- Hardmeier, I.; Aeberhard, N.; Qi, W.; Schoenbaechler, K.; Kraettli, H.; Hatt, J.-M.; Fraefel, C.; Kubacki, J. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS ONE 2021, 16, e0252534. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Members of the International Committee on the Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef]
- Wang, B.; Li, W.; Zhou, J.-H.; Li, B.; Zhang, W.; Yang, W.-H.; Pan, H.; Wang, L.-X.; Bock, C.T.; Shi, Z.-L.; et al. Chevrier’s Field Mouse (Apodemus chevrieri) and Père David’s Vole (Eothenomys melanogaster) in China Carry Orthohepeviruses that form Two Putative Novel Genotypes Within the Species Orthohepevirus C. Virol. Sin. 2018, 33, 44–58. [Google Scholar] [CrossRef]
- van Tong, H.; Hoan, N.X.; Wang, B.; Wedemeyer, H.; Bock, C.-T.; Velavan, T.P. Hepatitis E Virus Mutations: Functional and Clinical Relevance. EBioMedicine 2016, 11, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Yip, C.C.; Wu, S.; Chew, N.F.; Leung, K.H.; Chan, J.F.; Zhao, P.S.; Chan, W.M.; Poon, R.W.; Tsoi, H.; et al. Transmission of Rat Hepatitis E Virus Infection to Humans in Hong Kong: A Clinical and Epidemiological Analysis. Hepatology 2021, 73, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Heller, B.; Capuccino, J.M.V.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152, Erratum in Proc. Natl. Acad. Sci. USA 2017, 114, E4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Kumar, C.T.R.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Yadav, K.K.; Boley, P.A.; Fritts, Z.; Kenney, S.P. Ectopic Expression of Genotype 1 Hepatitis E Virus ORF4 Increases Genotype 3 HEV Viral Replication in Cell Culture. Viruses 2021, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Tanggis; Kobayashi, T.; Takahashi, M.; Jirintai, S.; Nishizawa, T.; Nagashima, S.; Nishiyama, T.; Kunita, S.; Hayama, E.; Tanaka, T.; et al. An analysis of two open reading frames (ORF3 and ORF4) of rat hepatitis E virus genome using its infectious cDNA clones with mutations in ORF3 or ORF4. Virus Res. 2018, 249, 16–30. [Google Scholar] [CrossRef]
- Hawkins, J.A.; Kaczmarek, M.E.; Müller, M.A.; Drosten, C.; Press, W.H.; Sawyer, S.L. A metaanalysis of bat phylogenetics and positive selection based on genomes and transcriptomes from 18 species. Proc. Natl. Acad. Sci. USA 2019, 116, 11351–11360. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Dong, N. Phylogenomic analyses of bat subordinal relationships based on transcriptome data. Sci. Rep. 2016, 6, 27726. [Google Scholar] [CrossRef] [Green Version]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Mollentze, N.; Streicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.Y.; Camens, A.B. Strong morphological support for the molecular evolutionary tree of placental mammals. J. Evol. Biol. 2009, 22, 2243–2257. [Google Scholar] [CrossRef]
- Sander, A.-L.; Corman, V.M.; Lukashev, A.N.; Drexler, J.F. Evolutionary Origins of Enteric Hepatitis Viruses. Cold Spring Harb. Perspect. Med. 2018, 8, a031690. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, R.; Forni, D.; Sironi, M. Mode and tempo of human hepatitis virus evolution. Comput. Struct. Biotechnol. J. 2019, 17, 1384–1395. [Google Scholar] [CrossRef]
- Brayne, A.B.; Dearlove, B.; Lester, J.; Pond, S.L.K.; Frost, S.D.W. Genotype-Specific Evolution of Hepatitis E Virus. J. Virol. 2017, 91, e02241-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero-Juarez, A.; Frias, M.; Perez, A.B.; Pineda, J.A.; Reina, G.; Fuentes-Lopez, A.; Freyre-Carrillo, C.; Ramirez-Arellano, E.; Alados, J.C.; Rivero, A. Orthohepevirus C infection as an emerging cause of acute hepatitis in Spain: First report in Europe. J. Hepatol. 2022. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Misra, V. Immune System Modulation and Viral Persistence in Bats: Understanding Viral Spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef] [Green Version]
- Scholz, J.; Falkenhagen, A.; Bock, C.-T.; Johne, R. Reverse genetics approaches for Hepatitis E virus and related viruses. Curr. Opin. Virol. 2020, 44, 121–128. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.-J. Hepatitis E Virus: Animal Models and Zoonosis. Annu. Rev. Anim. Biosci. 2019, 7, 427–448. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Genus | Species | Genotype | Host (Commonly) |
|---|---|---|---|---|---|
| Hepeviridae | Orthohepevirinae | Paslahepevirus | balayani | HEV-A1 | Human |
| HEV-A2 | Human | ||||
| HEV-A3 | Human, pig, wild boar, deer, rabbit | ||||
| HEV-A4 | Human, pig, wild boar | ||||
| HEV-A5 | Wild boar | ||||
| HEV-A6 | Wild boar | ||||
| HEV-A7 | Dromedary camel | ||||
| HEV-A8 | Bactrian camel | ||||
| alci | Moose | ||||
| Unclassified | Tree shrew (likely) | ||||
| Avihepevirus | magniiecur | Chicken, sparrow | |||
| egretti | Little egret | ||||
| Rocahepevirus | ratti | HEV-C1 | Rat, house shrew | ||
| HEV-C2 | Ferret | ||||
| HEV-C3 | Field mouse | ||||
| eothenomi | Vole | ||||
| Unclassified | Hamster | ||||
| Chirohepevirus | eptesici | Serotine, myotis bat | |||
| rhinolophi | Horseshoe bat | ||||
| desmodi | Vampire bat | ||||
| Parahepevirinae | Piscihepevirus | heenan | Trout |
| Family | Species | Common Name 1 | Sampling Site (Year) | Sample Source | Genomic Sequence (No.) | Reference |
|---|---|---|---|---|---|---|
| Vespertilionidae | Eptesicus serotinus | Serotine | Germany (2008) | Liver | Complete (1) | [36] |
| Eptesicus japonensis | Japanese short-tailed bat | Japan (2015) | Feces | Partial (2) | [55] | |
| Myotis bechsteinii | Bechstein’s myotis | Germany (2008) | Feces | Partial (1) | [36] | |
| Myotis daubentonii | Daubenton’s myotis | Germany (2008) | Feces | Partial (2) | [36] | |
| Myotis davidii | David’s myotis | China (2011) | Liver | Complete (1) | [54] | |
| Plecotus sacrimontis | Japanese long-eared bat | Japan (2015) | Feces | Partial (1) | [55] | |
| Pipistrellus nathusii | Nathusius’ pipistrelle | Switzerland (2019) | Feces | Nearly complete (1) 2 | [59] | |
| Phyllostomidae | Vampyrodes caraccioli | Great stripe-faced bat | Panama (2011) | Blood | Partial (1) | [36] |
| Desmodus rotundus | Vampire bat | Peru (2016) | Fecal swab | Complete (4) | [58] | |
| Mystacinidae | Mystacina tuberculata | New Zealand lesser short-tailed bat | New Zealand (2013) | Feces | Partial (1) | [56] |
| Rhinolophidae | Rhinolophus ferrumequinum | Greater horseshoe bat | China (2013) | Fecal swab | Complete (1) | [57] |
| Hipposideridae | Hipposideros abae | Aba roundleaf bat | Ghana (2009) | Feces | Partial (2) | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Yang, X.-L. Chirohepevirus from Bats: Insights into Hepatitis E Virus Diversity and Evolution. Viruses 2022, 14, 905. https://doi.org/10.3390/v14050905
Wang B, Yang X-L. Chirohepevirus from Bats: Insights into Hepatitis E Virus Diversity and Evolution. Viruses. 2022; 14(5):905. https://doi.org/10.3390/v14050905
Chicago/Turabian StyleWang, Bo, and Xing-Lou Yang. 2022. "Chirohepevirus from Bats: Insights into Hepatitis E Virus Diversity and Evolution" Viruses 14, no. 5: 905. https://doi.org/10.3390/v14050905
APA StyleWang, B., & Yang, X.-L. (2022). Chirohepevirus from Bats: Insights into Hepatitis E Virus Diversity and Evolution. Viruses, 14(5), 905. https://doi.org/10.3390/v14050905