
Ubiquitous Micro-Modular Homologies among Genomes from Viruses to Bacteria to Human Mitochondrial DNA: Platforms for Recombination during Evolution?




Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Problems Studied
3.2. Comparisons of Selected Segments of SARS-CoV-2 RNA with Segments of High Sequence Homologies in the Genomes of Diverse Viral and Non-Viral Families
- 1.
- Comparisons of the Wuhan SARS-CoV-2 nucleotide sequences to sequences from diverse virus families
- (i)
- Genomes of SARS-CoV-2 and human adenovirus types 2 and 12
- (ii)
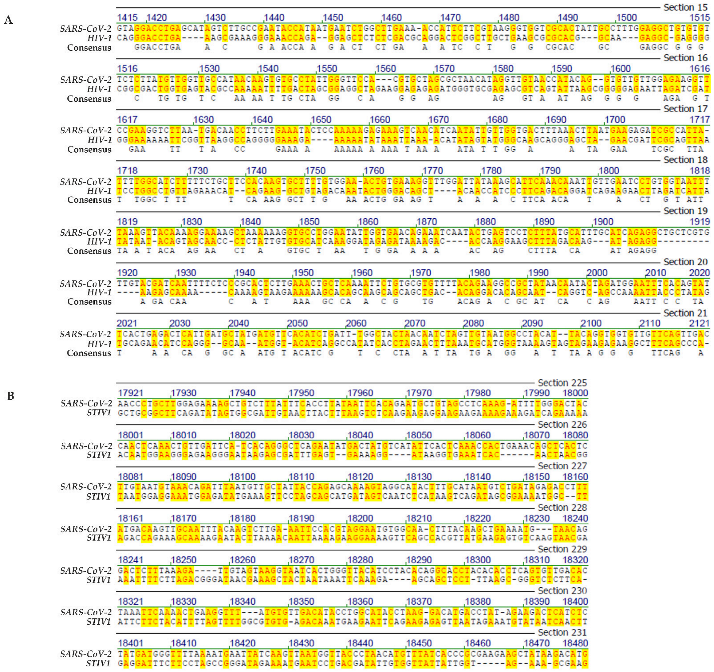
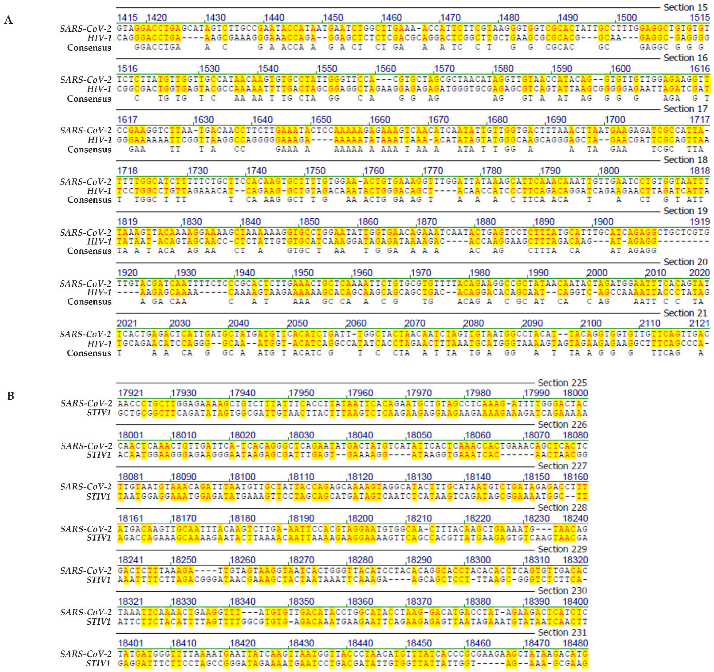
- Genome comparison: SARS-CoV-2 and human immunodeficiency virus-1 (HIV-1)
- (iii)
- Genome comparison: SARS-CoV-2 and of an archaea phage
- (iv)
- Genome comparison: SARS-CoV-2 and plant Alternaria brassicola endornavirus
- (v)
- Genome comparison: SARS-CoV-2 and insect Autographa californica nucleo- polyhedrovirus (AcNPV)
- 2.
- Homologies between SARS-CoV-2 RNA and proteobacterial and bacterial genomes
- (i)
- Genome comparison: SARS-CoV-2 and Candidatus Carsonella ruddii strain BT
- (ii)
- Genome comparison: SARS-CoV-2 and Escherichia coli strain K12
- 3.
- Genome comparison: SARS-CoV-2 RNA to human mitochondrial DNA.
- 4.
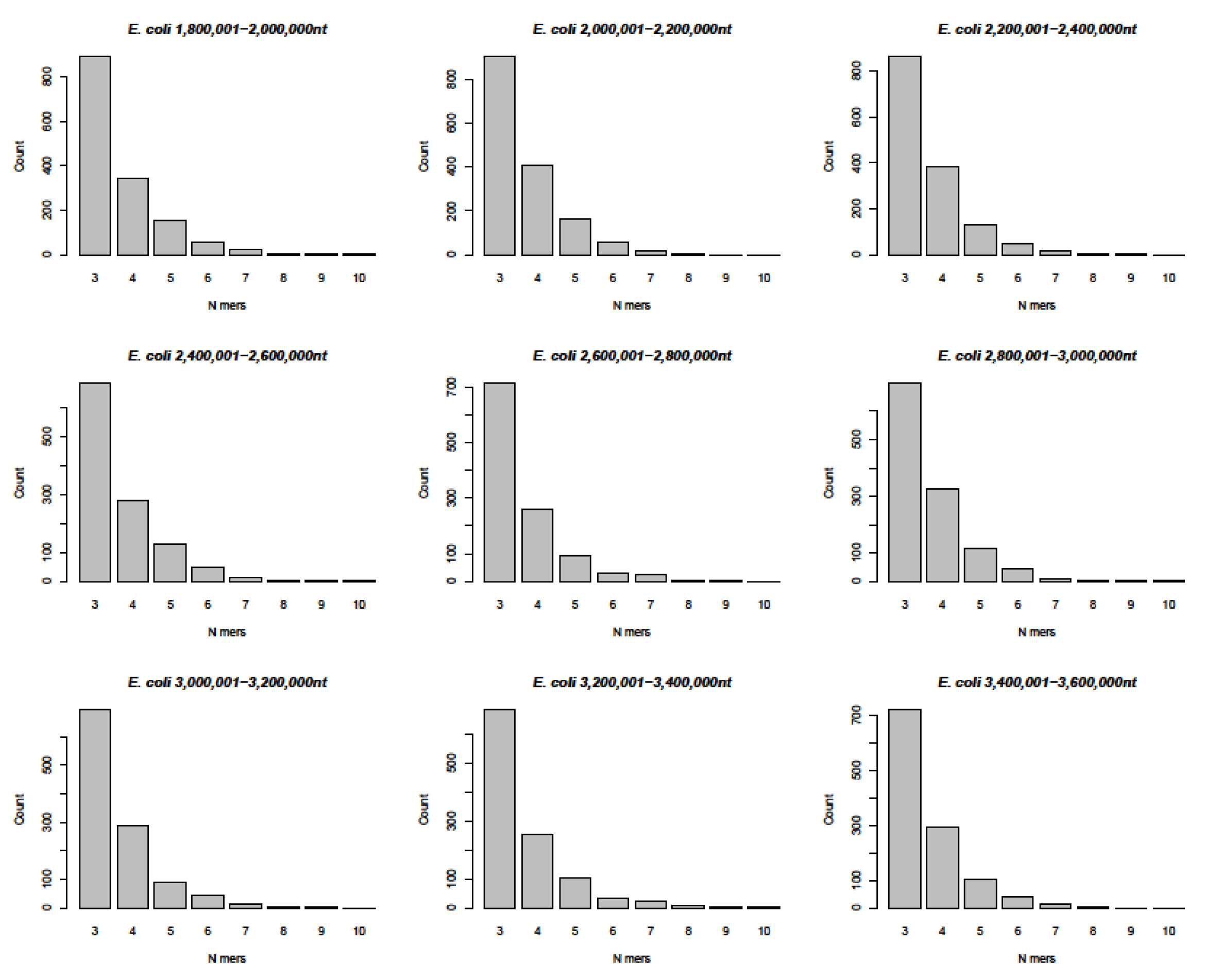
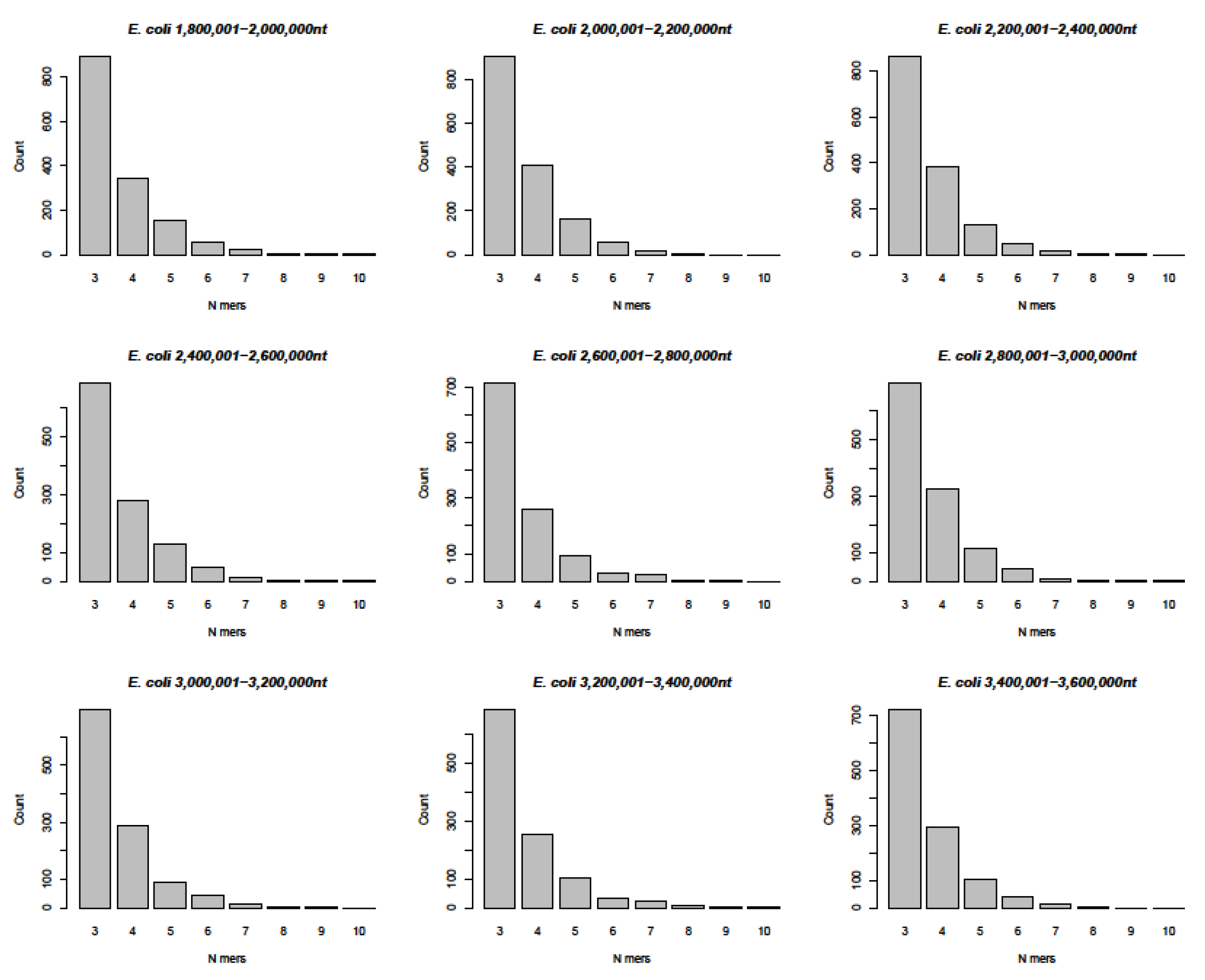
- Genome comparison: SARS-CoV-2 RNA to the entire Escherichia coli K12 chromosome of 4,641,652 nucleotide pairs
- 5.
- Statistical analysis (bar graphs) of inter-genomic micro-modular homology clusters of 3 to 9 and up to rarely occurring 13 nucleotides.
- 6.
- Control analyses with randomly computer-synthesized 29.9 kb DNA
4. Discussion and Outlook
- (i)
- Synopsis and interpretation of data
- (ii)
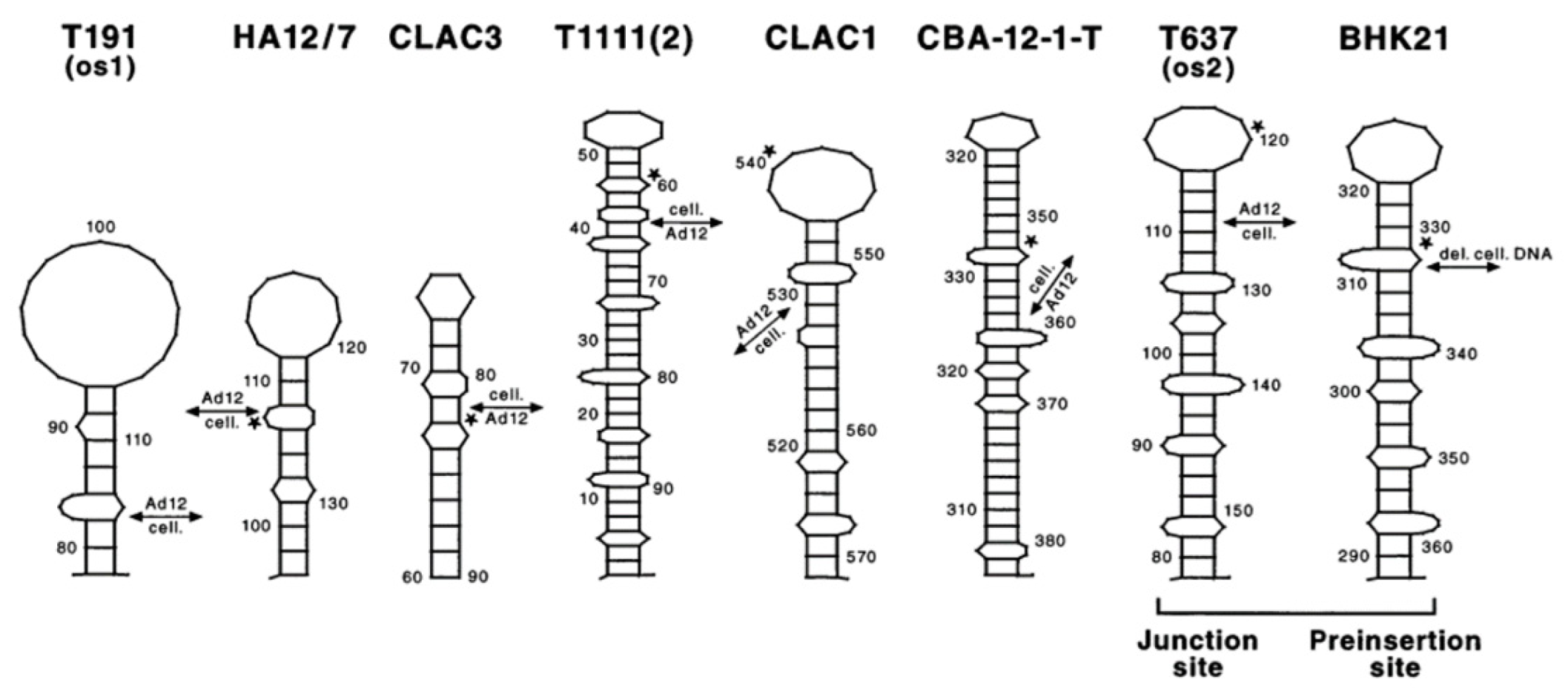
- Patch homologies and stem-loop structures between DNA molecules of very diverse origins
- (iii)
- The four-letter coding system and its selection during evolution—a hypothesis
5. Conclusions
- Genome structures of several organisms across the evolutionary spectrum were aligned with the SARS-CoV-2 genome sequence. Micro-modular homologies of n = 3 to n = 9, and up to n = 13 (rare case) were recorded to gain insights into generalities in the design of existing and evolving (SARS-CoV-2) genomes.
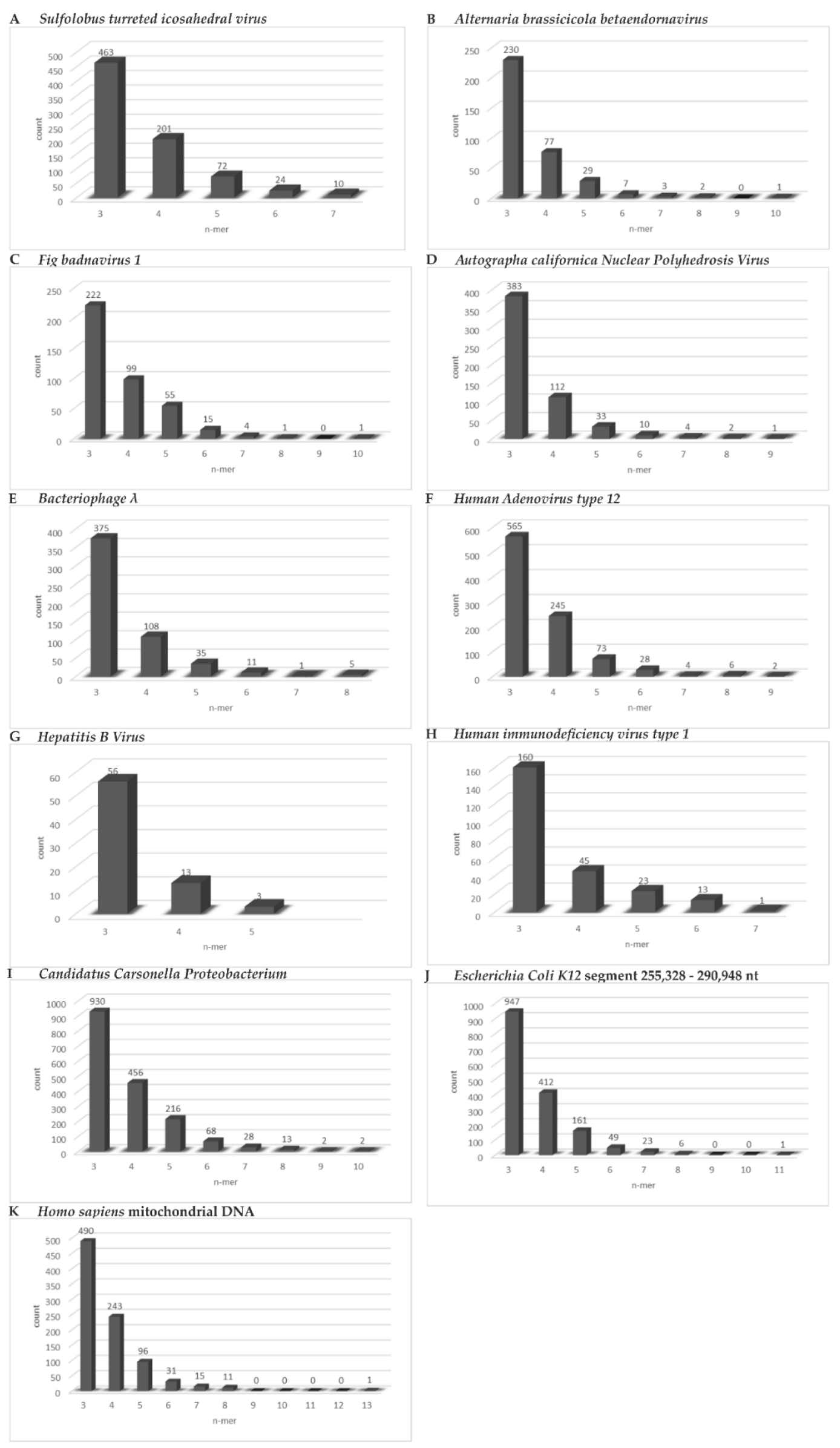
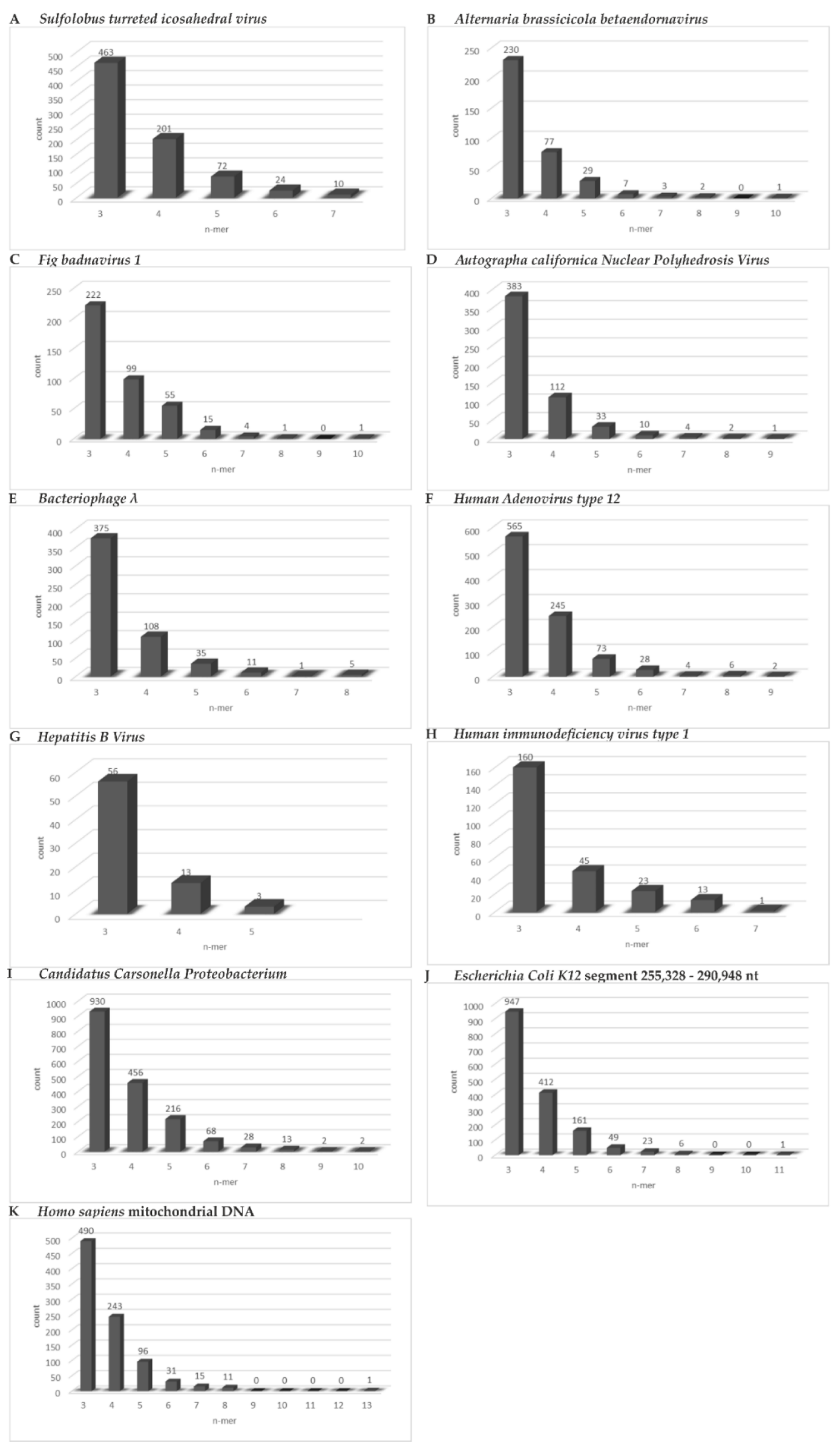
- We found abundant micro-modular identities across the spectrum of genomes and investigated and documented these data in the bar graphs depicted in Figure 6A–K.
- In a computer-aided sequence comparison, the sequence of the SARS-CoV-2 genome was matched to the entire 4,641,652 nt DNA sequence of Escherichia coli K12. This analysis yielded very similar results (Figure 6 and Figure S6A,B) with respect both to the numerical n = 3 to n = 9, and up to n > 10-mer arrays and to the complex patch pattern sequence homologies, reminiscent of those described in integrative recombination reactions [11,12,13,14,15,16].
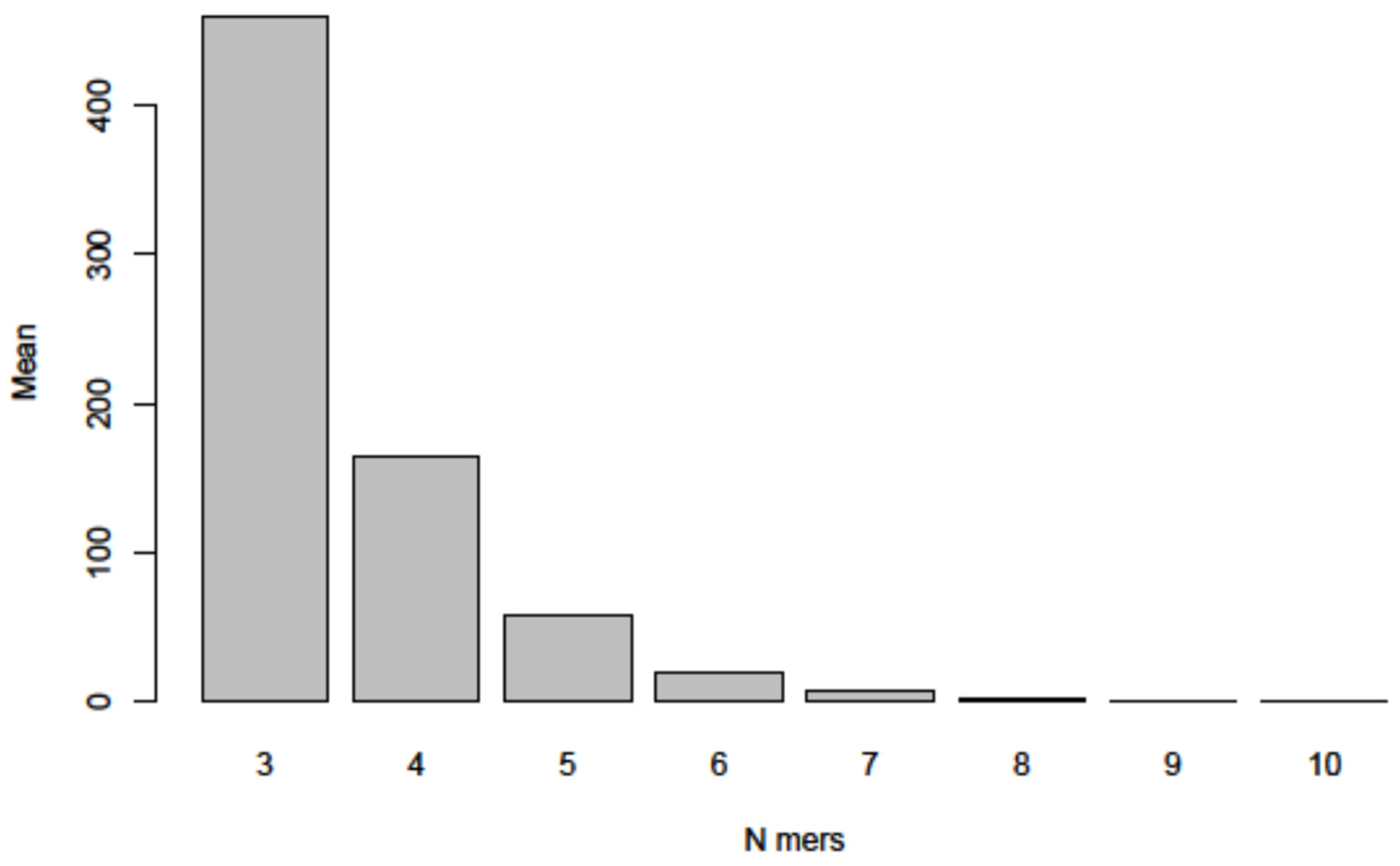
- In an extensive control project, we subjected 1000 randomly generated nucleotide sequences of the length and base composition of the SARS-CoV-2 genome to the same type of analyses for micro-modular sequence identities and found very similar patterns of micro-modular homologies, now presented in Figure 8.
- The four letter A, C, G, T genetic encoding strategy has proven exceptionally successful during 4 billion years of evolution. We submit that the system-inherent capability to generate a vast abundance of sequence and pattern identities alternating with non-identities to the tune of “patchy homologies” has provided opportunities for biochemically meaningful platforms for DNA-protein interactions and for (integrative) recombination reactions. In the course of evolutionary time periods, this system has facilitated the annealing of nucleotide sequences required for repeated extensions of initially smaller nucleotide sequences to the real genomes of enormous lengths that exist today. Moreover, the actually existing patterns (see alignments in Figures S1–S4) offer unidentified recognition signals for recombination events between otherwise unrelated genomes.
- It will take systematic investigations to explore the possibility of whether these mechanisms might have served also in the generation and selection of variants of newly emerging pathogenic viruses like SARS-CoV-2 and others.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L. Geneva Centre for Emerging Viral Diseases. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Yusof, W.; Irekeola, A.A.; Wada, Y.; Engku Abd Rahman, E.N.S.; Ahmed, N.; Musa, N.; Khalid, M.F.; Rahman, Z.A.; Hassan, R.; Yusof, N.Y.; et al. A Global Mutational Profile of SARS-CoV-2: A Systematic Review and Meta-Analysis of 368,316 COVID-19 Patients. Life 2021, 11, 1224. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Ramirez, C.; Doerfler, W. Signal hotspot mutations in SARS-CoV-2 genomes evolve as the virus spreads and actively replicates in different parts of the world. Virus Res. 2020, 289, 198170. [Google Scholar] [CrossRef]
- Weber, S.; Ramirez, C.M.; Weiser, B.; Burger, H.; Doerfler, W. SARS-CoV-2 worldwide replication drives rapid rise and selection of mutations across the viral genome: A time-course study—potential challenge for vaccines and therapies. EMBO Mol. Med. 2021, 13, e14062. [Google Scholar] [CrossRef]
- Anant, S. Davidson NO Molecular mechanisms of apolipoprotein B mRNA editing. Curr. Opin. Lipidol. 2001, 12, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Yin, W.; Xu, H.E. RNA-dependent RNA polymerase: Structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Nakabachi, A.; Yamashita, A.; Toh, H.; Ishikawa, H.; Dunbar, H.E.; Moran, N.A.; Hattori, M. The 160-kilobase genome of the bacterial endosymbiont. Carsonella Sci. 2006, 314, 267. [Google Scholar] [CrossRef] [Green Version]
- Botchan, M.; Stringer, J.; Mitchison, T.; Sambrook, J. Integration and excision of SV40 DNA from the chromosome of a transformed cell. Cell 1980, 20, 143–152. [Google Scholar] [CrossRef]
- Stabel, S.; Doerfler, W.; Friis, R.R. Integration sites of adenovirus type 12 DNA in transformed hamster cells and hamster tumor cells. J. Virol. 1980, 36, 22–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deuring, R.; Winterhoff, U.; Tamanoi, F.; Stabel, S.; Doerfler, W. Site of linkage between adenovirus type 12 and cell DNAs in hamster tumour line CLAC3. Nature 1981, 293, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Gahlmann, R.; Leisten, R.; Vardimon, L.; Doerfler, W. Patch homologies and the integration of adenovirus DNA in mammalian cells. EMBO J. 1982, 1, 1101–1104. [Google Scholar] [CrossRef]
- Doerfler, W.; Gahlmann, R.; Stabel, S.; Deuring, R.; Lichtenberg, U.; Schulz, M.; Eick, D.; Leisten, R. On the mechanism of recombination between adenoviral and cellular DNAs: The structure of junction sites. Curr. Top. Microbiol. Immunol. 1984, 109, 193–228. [Google Scholar] [CrossRef]
- Knoblauch, M.; Schröer, J.; Schmitz, B.; Doerfler, W. The structure of adenovirus type 12 DNA integration sites in the hamster cell genome. J. Virol. 1996, 70, 3788–3796. [Google Scholar] [CrossRef] [Green Version]
- Doerfler, W. 2000 Weinheim, New York, Chichester, Brisbane, Singapore, Toronto; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens. N. Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Ananth, S.; Morath, K.; Trautz, B.; Tibroni, N.; Shytaj, I.L.; Obermaier, B.; Stolp, B.; Lusic, M.; Fackler, O.T. Multifunctional Roles of the N-Terminal Region of HIV-1SF2Nef Are Mediated by Three Independent Protein Interaction Sites. J. Virol. 2019, 94, e01398-19. [Google Scholar] [CrossRef]
- Mandala, W.L.; Liu, M.K.P. SARS-CoV-2 and HIV-1: Should HIV-1-Infected Individuals in Sub-Saharan Africa Be Considered a Priority Group for the COVID-19 Vaccines? Front. Immunol. 2021, 12, 797117. [Google Scholar] [CrossRef]
- Nablbantoglu, J.; Miles, C.; Meuth, M. Insertion of unique and repetitive DNA fragments into the aprt locus of hamster cells. J. Mol. Biol. 1988, 200, 449–459. [Google Scholar] [CrossRef]
- Hyrien, O.; Debatisse, M.; Buttin, G.; Vincent, B.R.d. A hotspot for novel amplification joints in a mosaic of Alu-like repeats and palindromic A1T rich DNA. EMBO J. 1987, 6, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Bullock, P.; Forrester, W.; Botchan, M. DNA sequence studies of simian virus 40 chromosomal excision and integration in rat cells. J. Mol. Biol. 1984, 174, 55–84. [Google Scholar] [CrossRef]
- Romani, M.; de Ambrosis, A.; Alhadeff, B.; Purrello, M.; Gluzman, Y.; Siniscalco, M. Preferential integration of the Ad5/SV40 hybrid virus at the highly recombingenic human chromosomal site 1p36. Gene 1990, 95, 231–241. [Google Scholar] [CrossRef]
- Delbrück, M. A physicist’s renewed look at biology: Twenty years later. Science 1970, 168, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; Macleod, C.M.; McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Kaiser, A.D. Structure and base sequence in the cohesive ends of bacteriophage lambda DNA. J. Mol. Biol. 1968, 35, 523–537. [Google Scholar] [CrossRef]
- Jaeger, J.A.; Turner, D.H.; Zuker, M. Improved predictions of secondary structures for RNA. Proc. Natl. Acad. Sci. USA 1989, 86, 7706–7710. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomes | Overall Homology | In Selected Segments |
|---|---|---|
| Archaea Viruses | ||
| Acidianus rod-shaped virus 1 | 41.4% | 46.2% |
| Nitrosopumilus spindle-shaped virus | 45.8% | 47% |
| Sulfolobus turreted icosahedral virus | 30.3% | 45.5% |
| Bacteriophages | ||
| Escherichia virus lambda (λ) | 30.6% | 45.3% |
| Escherichia virus T4 | 9% | 46.4% |
| Escherichia virus mu | 39.7% | 44.2% |
| Plant Viruses | ||
| Alternaria brassicicola endornavirus | 13.1% | 44.5% |
| Carrot mottle mimic umbravirus (ds-RNA) | 5.9% | 45% |
| Fig badnavirus 1 | 11.8% | 45.5% |
| Insect Virus | ||
| Autographa californica Nuclear Polyhedrosis Virus | 11.2% | 45.6% |
| Human Viruses | ||
| Adenovirus type 2 | 38.4% | 43.2% |
| Adenovirus type 5 | 40% | 43.4% |
| Adenovirus type 12 | 43.2% | 44.9% |
| Hepatitis B Virus | 5.4% | 45.7% |
| Human Immunodeficiency Virus 1 | 16.6% | 45.8% |
| Bacterial Genomes (Segments) | ||
| Escherichia coli K12 (129,062–161,212 nt) | 45.2% | |
| Candidatus Carsonella ruddii strain BT | 8.5% | 46% |
| Mitochondrial DNA—homo sapiens | 27.3% | 45.3% |
| Random DNA | Homology | |
|---|---|---|
| Random DNA 1 versus SARS-CoV-2 | 44.7% | Average: 44.5% |
| Random DNA 2 versus SARS-CoV-2 | 44.8% | |
| Random DNA 3 versus SARS-CoV-2 | 44.4% | |
| Random DNA 4 versus SARS-CoV-2 | 44.5% | |
| Random DNA 5 versus SARS-CoV-2 | 44.3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, S.; Ramirez, C.M.; Doerfler, W. Ubiquitous Micro-Modular Homologies among Genomes from Viruses to Bacteria to Human Mitochondrial DNA: Platforms for Recombination during Evolution? Viruses 2022, 14, 885. https://doi.org/10.3390/v14050885
Weber S, Ramirez CM, Doerfler W. Ubiquitous Micro-Modular Homologies among Genomes from Viruses to Bacteria to Human Mitochondrial DNA: Platforms for Recombination during Evolution? Viruses. 2022; 14(5):885. https://doi.org/10.3390/v14050885
Chicago/Turabian StyleWeber, Stefanie, Christina M. Ramirez, and Walter Doerfler. 2022. "Ubiquitous Micro-Modular Homologies among Genomes from Viruses to Bacteria to Human Mitochondrial DNA: Platforms for Recombination during Evolution?" Viruses 14, no. 5: 885. https://doi.org/10.3390/v14050885
APA StyleWeber, S., Ramirez, C. M., & Doerfler, W. (2022). Ubiquitous Micro-Modular Homologies among Genomes from Viruses to Bacteria to Human Mitochondrial DNA: Platforms for Recombination during Evolution? Viruses, 14(5), 885. https://doi.org/10.3390/v14050885