
Recent Zoonotic Spillover and Tropism Shift of a Canine Coronavirus Is Associated with Relaxed Selection and Putative Loss of Function in NTD Subdomain of Spike Protein

 , , , , and
, , , , and 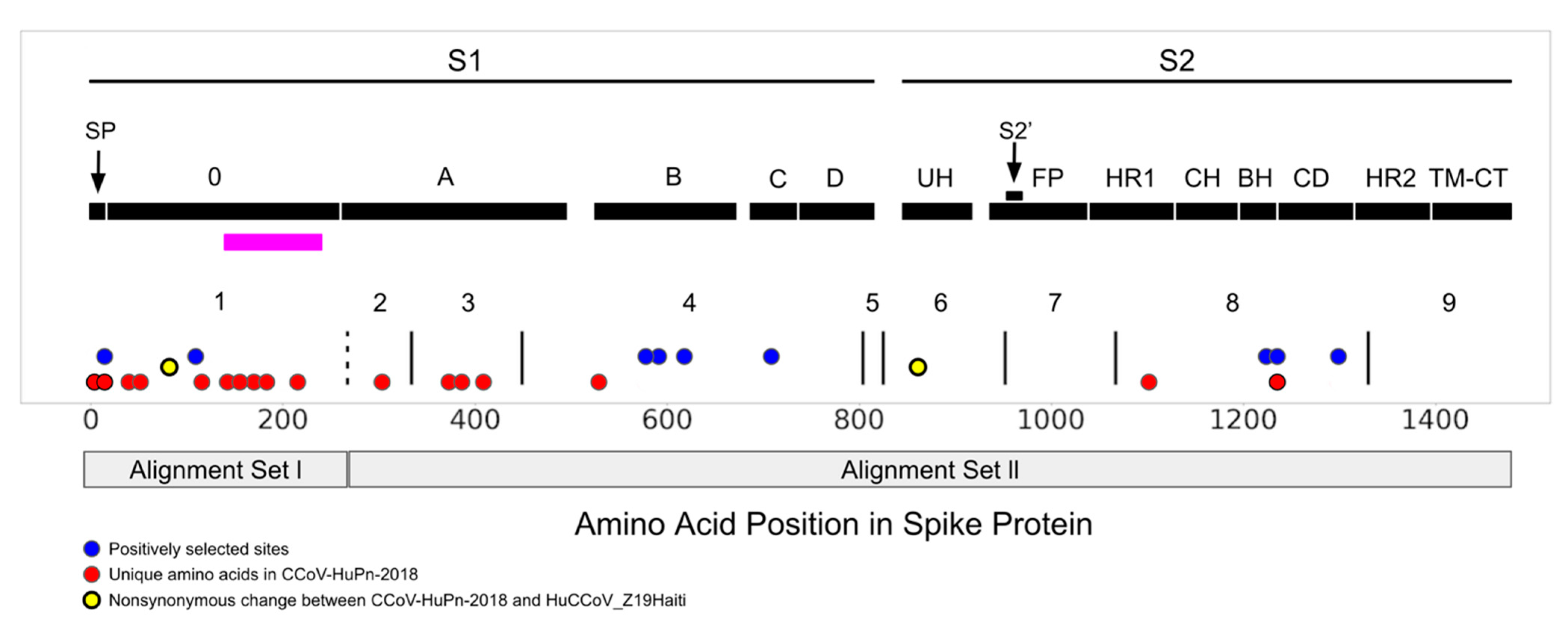{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequences and Alignments
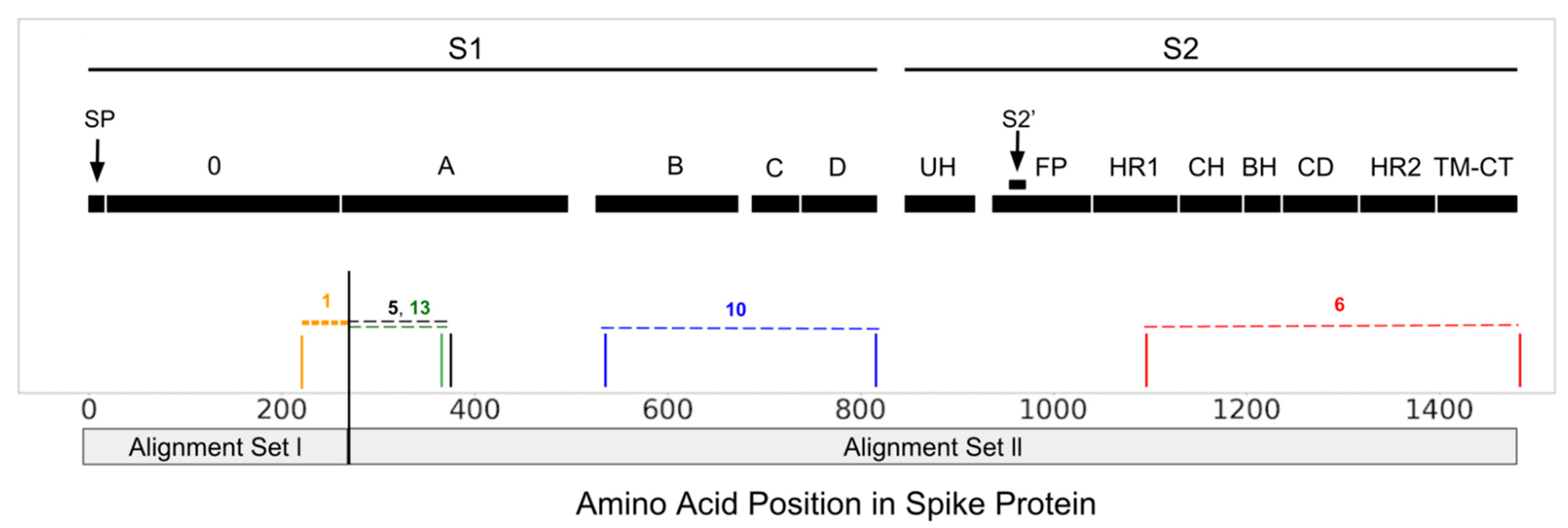
2.2. Recombination, Positive Selection, Temporal Dating
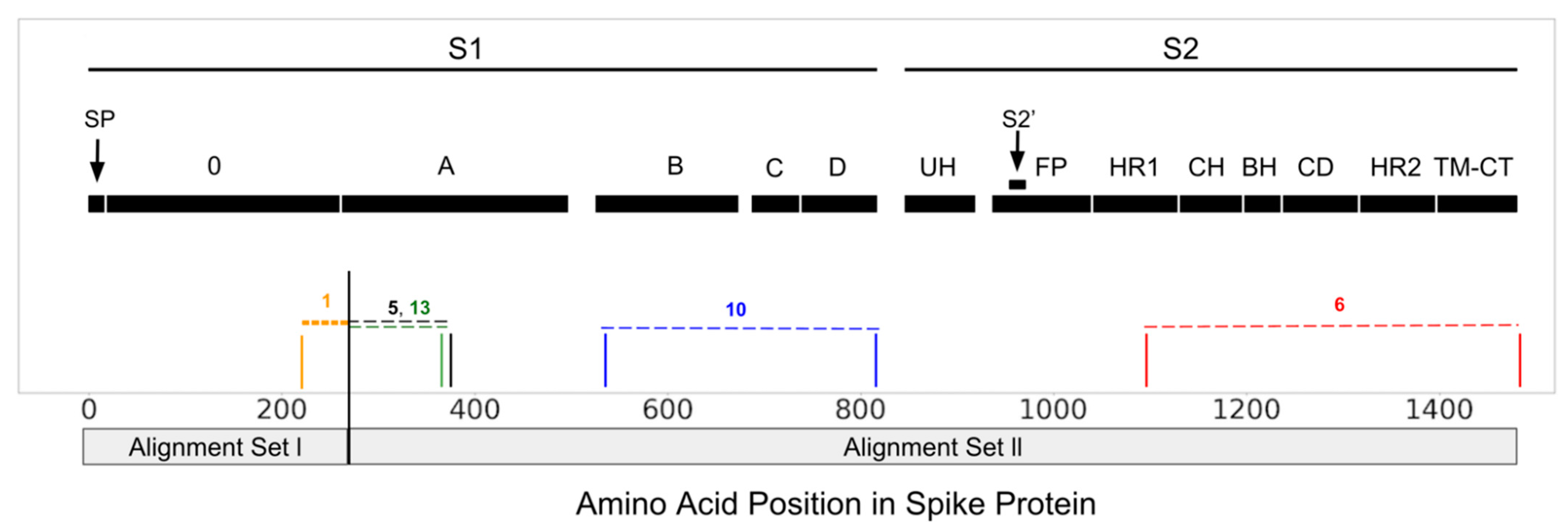
3. Results
3.1. Recombination and Temporal Dating
3.2. Selection Pressure
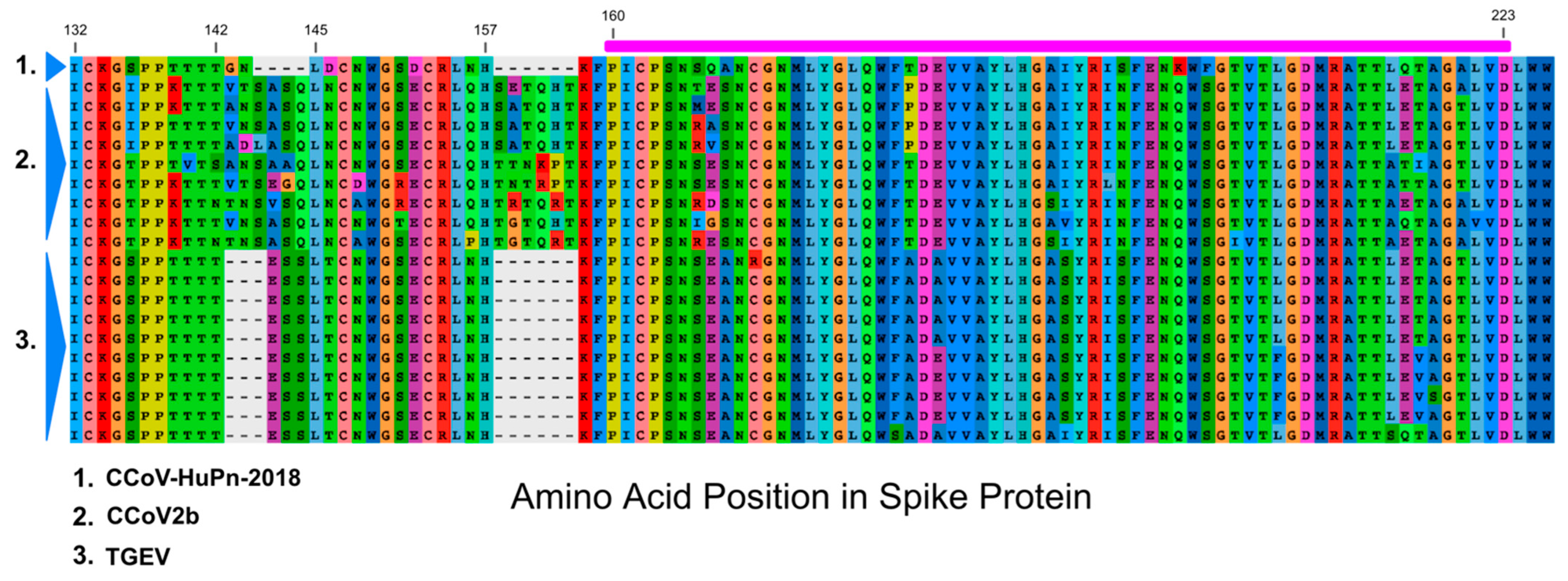
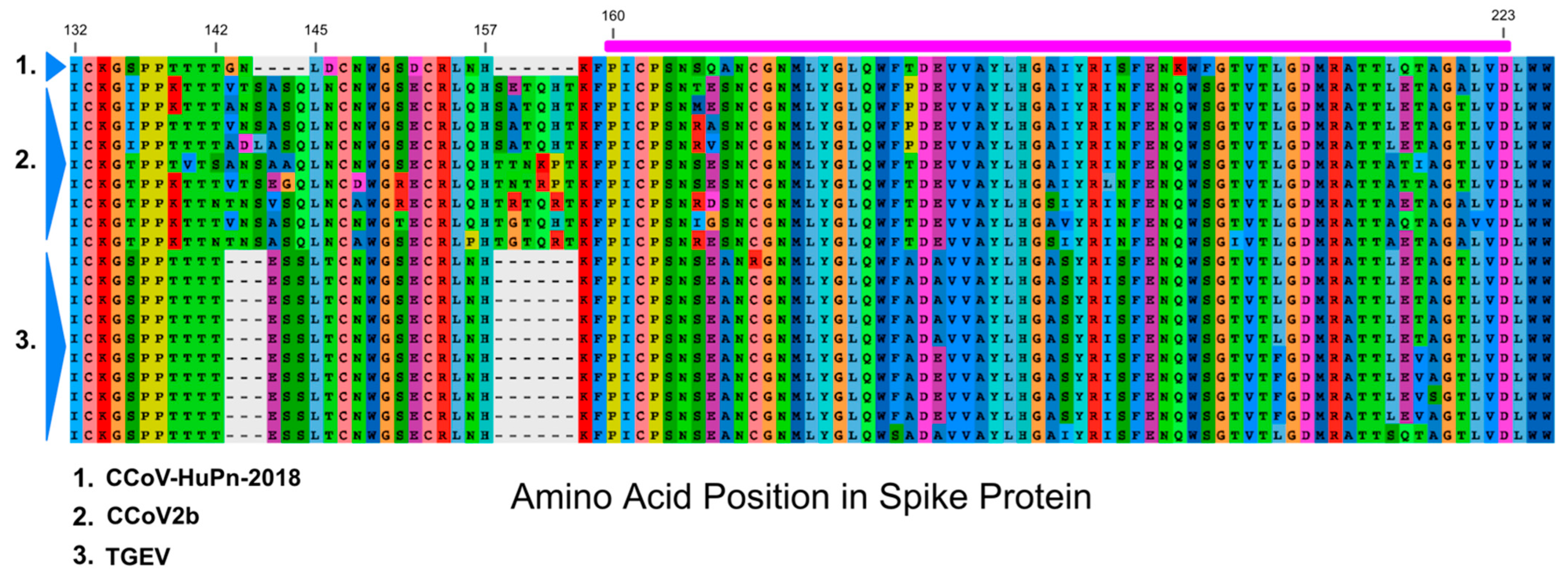
3.3. Sialic Acid Binding Region
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.H.; Lee, J.S.Y.; Saif, L.J.; Gray, G.C. Novel Canine Coronavirus Isolated from a Hospitalized Patient With Pneumonia in East Malaysia. Clin. Infect. Dis. 2022, 74, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Blohm, G.M.; Alam, M.M.; Iovine, N.M.; Salemi, M.; Mavian, C.; Morris, J.G. Isolation of a novel recombinant Canine Coronavirus from a visitor to Haiti: Further evidence of transmission of Coronaviruses of zoonotic origin to humans. Clin. Infect. Dis. 2021, ciab924. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Walls, A.C.; Joshi, A.; Park, Y.-J.; Eguia, R.T.; Stevens-Ayers, T.; Boeckh, M.J.; Telenti, A.; Lanzavecchia, A.; Corti, D.; et al. Structure, receptor recognition and antigenicity of the human coronavirus CCoV-HuPn-2018 spike glycoprotein. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Jaimes, J.A.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2021, 45, fuaa057. [Google Scholar] [CrossRef]
- Reguera, J.; Santiago, C.; Mudgal, G.; Ordoño, D.; Enjuanes, L.; Casasnovas, J.M. Structural bases of coronavirus attachment to host aminopeptidase N and its inhibition by neutralizing antibodies. PLoS Pathog. 2012, 8, e1002859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.H.; Tomlinson, A.C.; Zhou, D.; Satkunarajah, M.; Chen, K.; Sharon, C.; Desforges, M.; Talbot, P.J.; Rini, J.M. Receptor-binding loops in alphacoronavirus adaptation and evolution. Nat. Commun. 2017, 8, 1735. [Google Scholar] [CrossRef]
- Tresnan, D.B.; Levis, R.; Holmes, K.V. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup l. J. Virol. 1996, 70, 8669–8674. [Google Scholar] [CrossRef] [Green Version]
- Wesley, R.D. The S gene of canine coronavirus, strain UCD-1, is more closely related to the S gene of transmissible gastroenteritis virus than to that of feline infectious peritonitis virus. Virus Res. 1999, 61, 145–152. [Google Scholar] [CrossRef]
- Schultze, B.; Krempl, C.; Ballesteros, M.L.; Shaw, L.; Schauer, R.; Enjuanes, L.; Herrler, G. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (N-glycolylneuraminic acid) binding activity. J. Virol. 1996, 170, 5634–5637. [Google Scholar] [CrossRef] [Green Version]
- Krempl, C.; Schultze, B.; Laude, H.; Herrler, G. Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J. Virol. 1997, 71, 3285–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulswit, R.J.; de Haan, C.A.; Bosch, B.J. Coronavirus spike protein and tropism changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Markova-Raina, P.; Petrov, D. High sensitivity to aligner and high rate of false positives in the estimates of positive selection in the 12 Drosophila genomes. Genome Res. 2011, 21, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020, 7, veaa087. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold-Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertheim, J.O.; Murrell, B.; Smith, M.D.; Kosakovsky Pond, S.L.; Scheffler, K. RELAX: Detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 2015, 32, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Duchêne, S.; Duchêne, D.; Holmes, E.C.; Ho, S.Y. The performance of the Date-Randomization Test in phylogenetic analyses of time-structured virus data. Mol. Biol. Evol. 2015, 32, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Rieux, A.; Khatchikian, C.E. Tipdatingbeast: An r package to assist the implementation of phylogenetic tip-dating tests using beast. Mol. Ecol. Resour. 2017, 17, 608–613. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Philippe, H. Computing Bayes factors using thermodynamic integration. Syst. Biol. 2006, 55, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Krempl, C.; Ballesteros, M.L.; Zimmer, G.; Enjuanes, L.; Klenk, H.D.; Herrler, G. Characterization of the sialic acid binding activity of transmissible gastroenteritis coronavirus by analysis of haemagglutination-deficient mutants. J. Gen. Virol. 2011, 81, 489–496. [Google Scholar] [CrossRef]
- McKeirnan, A.J.; Evermann, J.F.; Hargis, A.; Ott, R.L. Isolation of feline coronaviruses from two cats with diverse disease manifestations. Feline Pract. 1981, 11, 16–20. [Google Scholar]
- Yang, T.J.; Chang, Y.C.; Ko, T.P.; Draczkowski, P.; Chien, Y.C.; Chang, Y.C.; Wu, K.P.; Khoo, K.H.; Chang, H.W.; Hsu, S.D. Cryo-EM analysis of a feline coronavirus spike protein reveals a unique structure and camouflaging glycans. Proc. Natl. Acad. Sci. USA 2020, 117, 1438–1446. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef] [Green Version]
- Decaro, N.; Mari, V.; Elia, G.; Lanave, G.; Dowgier, G.; Colaianni, M.L.; Martella, V.; Buonavoglia, C. Full-length genome analysis of canine coronavirus type I. Virus Res. 2015, 210, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.L. The role of cell surface sialic acids for SARS-CoV-2 infection. Glycobiology 2021, 31, 1245–1253. [Google Scholar] [CrossRef]
- Liu, C.; Tang, J.; Ma, Y.; Liang, X.; Yang, Y.; Peng, G.; Qi, Q.; Jiang, S.; Li, J.; Du, L.; et al. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J. Virol. 2015, 89, 6121–6125. [Google Scholar] [CrossRef] [Green Version]
- Kuchipudi, S.V.; Nelli, R.K.; Gontu, A.; Satyakumar, R.; Surendran Nair, M.; Subbiah, M. Sialic acid receptors: The key to solving the enigma of zoonotic virus spillover. Viruses 2021, 13, 262. [Google Scholar] [CrossRef]
- Rasschaert, D.; Duarte, M.; Laude, H. Porcine respiratory coronavirus differs from transmissible gastroenteritis virus by a few genomic deletions. J. Gen. Virol. 1990, 171, 2599–2607. [Google Scholar] [CrossRef]
- Sanchez, C.M.; Pascual-Iglesias, A.; Sola, I.; Zuñiga, S.; Enjuanes, L. Minimum determinants of Transmissible Gastroenteritis Virus enteric tropism are located in the N-terminus of spike protein. Pathogens 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- George, S.; Chattopadhyay, P.A.; Gagnon, J.; Timalsina, S.; Singh, P.; Vydyam, P.; Munshi, M.; Chiu, J.E.; Renard, I.; Harden, C.A.; et al. Evidence for SARS-CoV-2 Spike Protein in the Urine of COVID-19 Patients. Kidney360 2021, 2, 924–936. [Google Scholar] [CrossRef]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [Green Version]
- Yolitz, J.; Schwing, C.; Chang, J.; Van Ryk, D.; Nawaz, F.; Wei, D.; Cicala, C.; Arthos, J.; Fauci, A.S. Signal peptide of HIV envelope protein impacts glycosylation and antigenicity of gp120. Proc. Natl. Acad. Sci. USA 2018, 115, 2443–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozzi, A.; Biolatti, M.; Cagliani, R.; Forni, D.; Dell’Oste, V.; Pontremoli, C.; Vantaggiato, C.; Pozzoli, U.; Clerici, M.; Landolfo, S.; et al. Past and ongoing adaptation of human cytomegalovirus to its host. PLoS Pathog. 2020, 16, e1008476. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zehr, J.D.; Pond, S.L.K.; Martin, D.P.; Ceres, K.; Whittaker, G.R.; Millet, J.K.; Goodman, L.B.; Stanhope, M.J. Recent Zoonotic Spillover and Tropism Shift of a Canine Coronavirus Is Associated with Relaxed Selection and Putative Loss of Function in NTD Subdomain of Spike Protein. Viruses 2022, 14, 853. https://doi.org/10.3390/v14050853
Zehr JD, Pond SLK, Martin DP, Ceres K, Whittaker GR, Millet JK, Goodman LB, Stanhope MJ. Recent Zoonotic Spillover and Tropism Shift of a Canine Coronavirus Is Associated with Relaxed Selection and Putative Loss of Function in NTD Subdomain of Spike Protein. Viruses. 2022; 14(5):853. https://doi.org/10.3390/v14050853
Chicago/Turabian StyleZehr, Jordan D., Sergei L. Kosakovsky Pond, Darren P. Martin, Kristina Ceres, Gary R. Whittaker, Jean K. Millet, Laura B. Goodman, and Michael J. Stanhope. 2022. "Recent Zoonotic Spillover and Tropism Shift of a Canine Coronavirus Is Associated with Relaxed Selection and Putative Loss of Function in NTD Subdomain of Spike Protein" Viruses 14, no. 5: 853. https://doi.org/10.3390/v14050853
APA StyleZehr, J. D., Pond, S. L. K., Martin, D. P., Ceres, K., Whittaker, G. R., Millet, J. K., Goodman, L. B., & Stanhope, M. J. (2022). Recent Zoonotic Spillover and Tropism Shift of a Canine Coronavirus Is Associated with Relaxed Selection and Putative Loss of Function in NTD Subdomain of Spike Protein. Viruses, 14(5), 853. https://doi.org/10.3390/v14050853