
Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
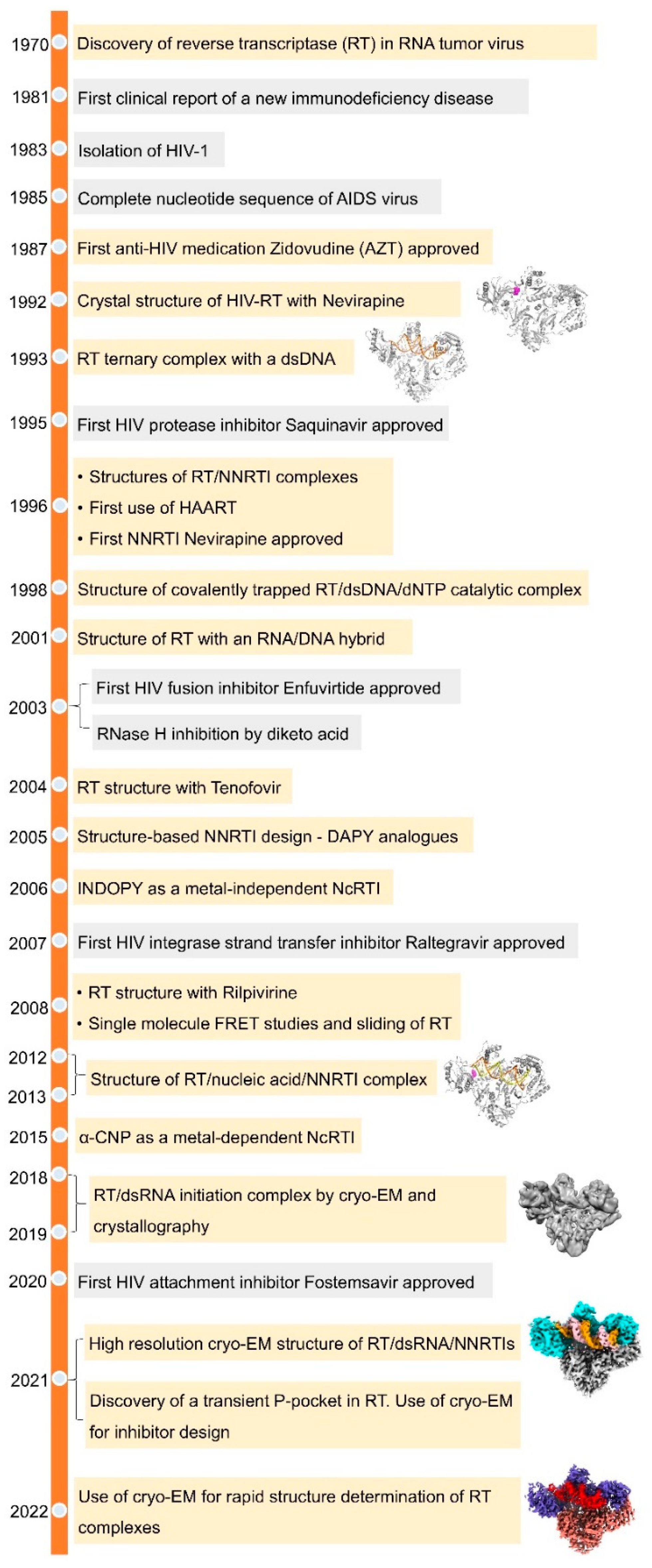
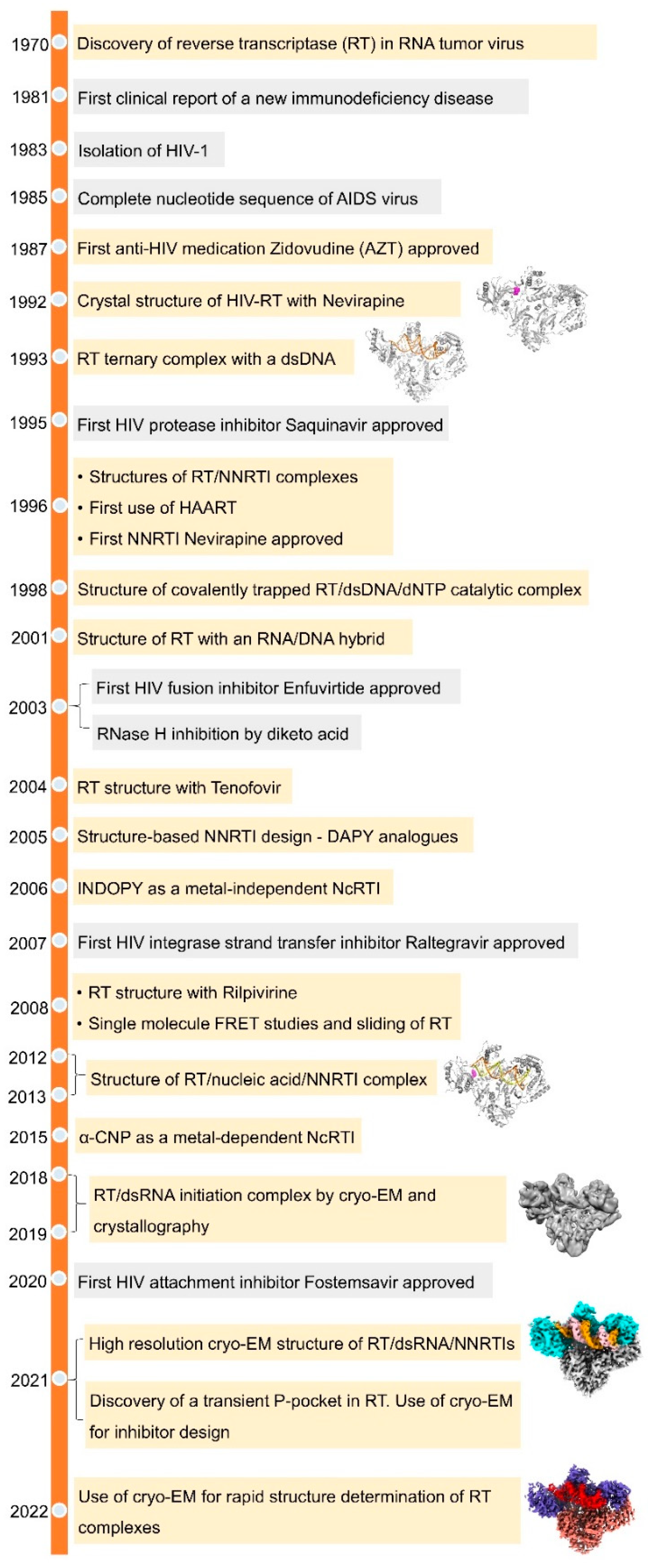
:1. Discovery of Reverse Transcriptase and Its HIV Connection
2. HIV Replication Cycle and RT
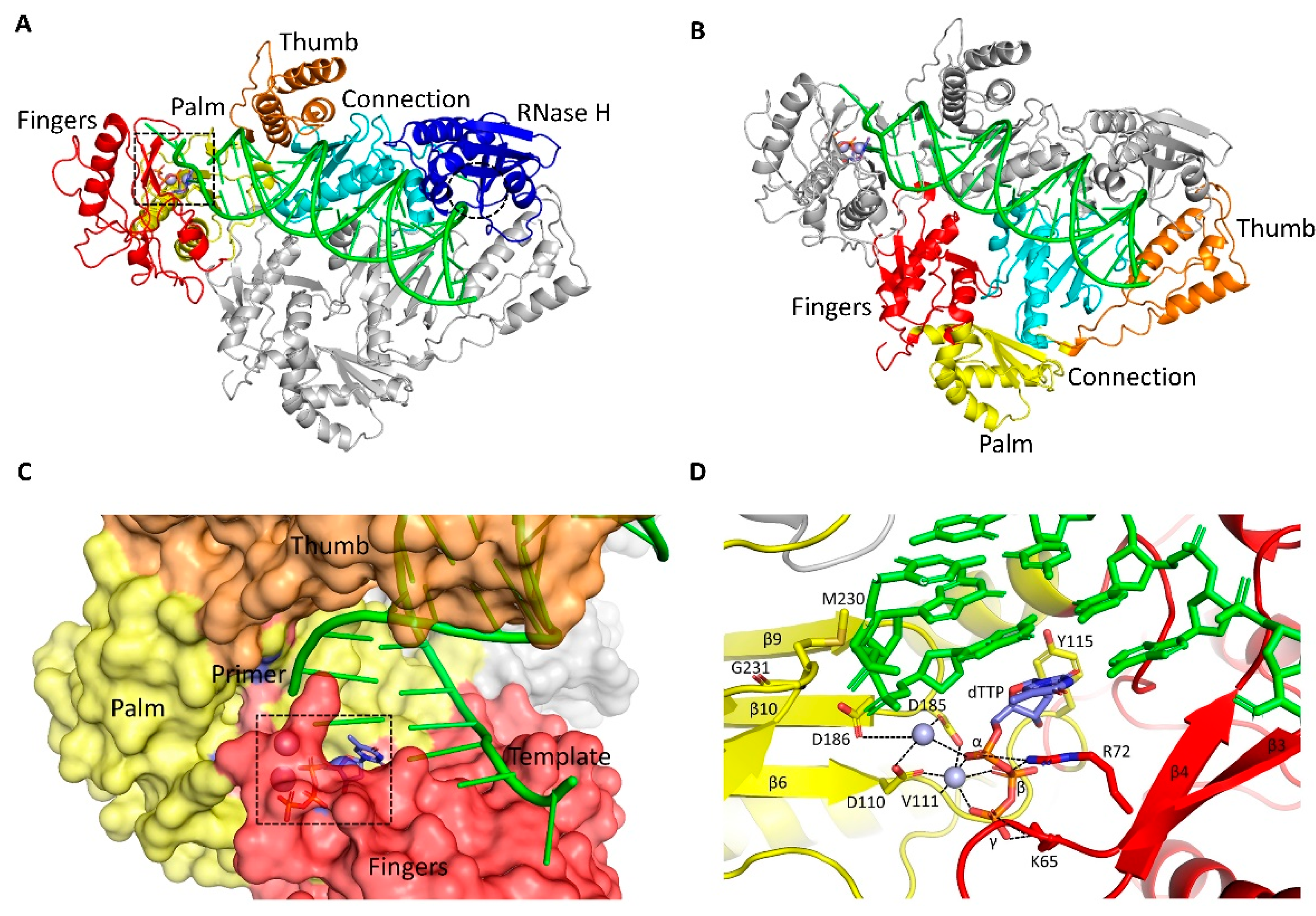
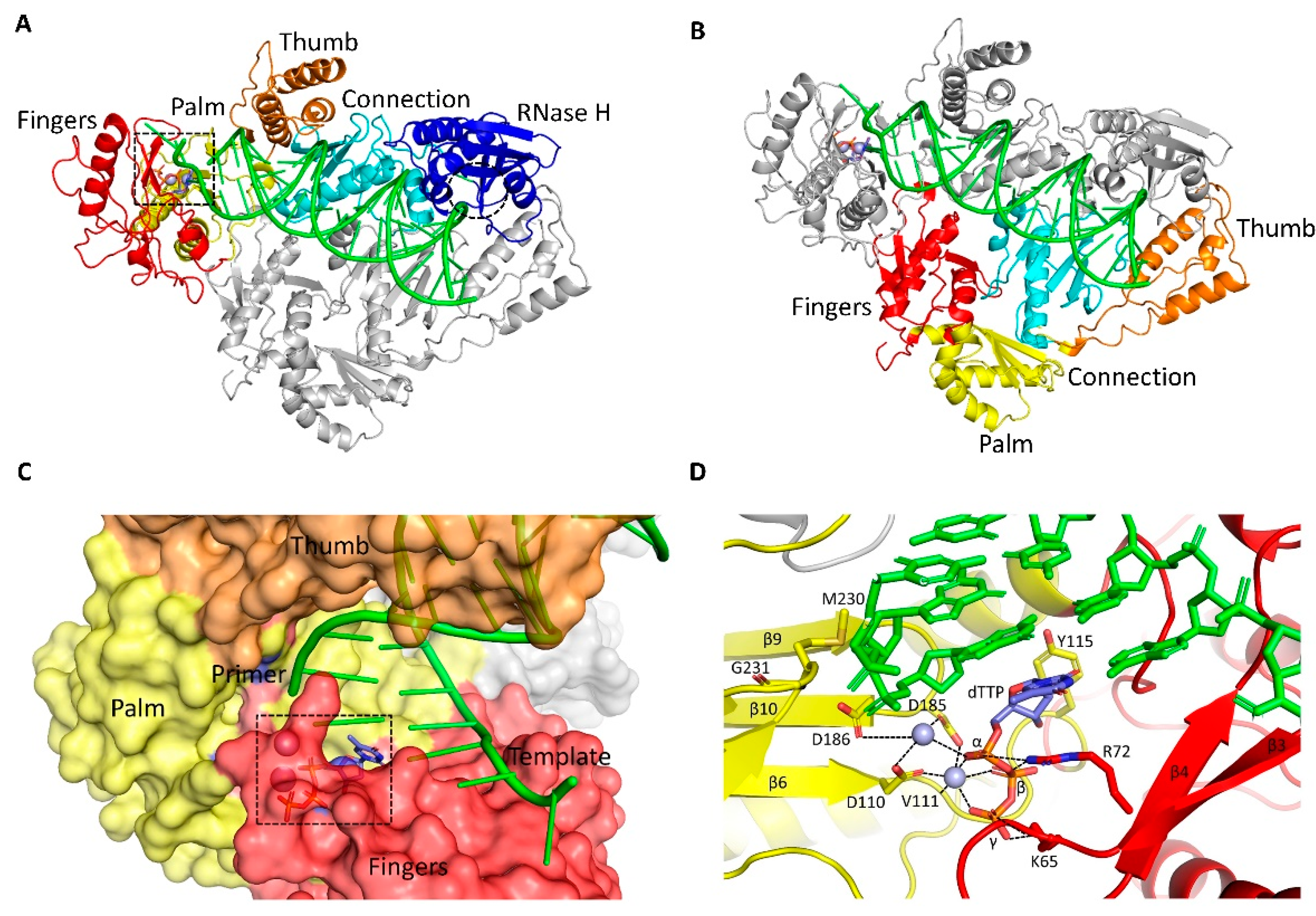
3. Structures of RT
4. Drugs Targeting RT
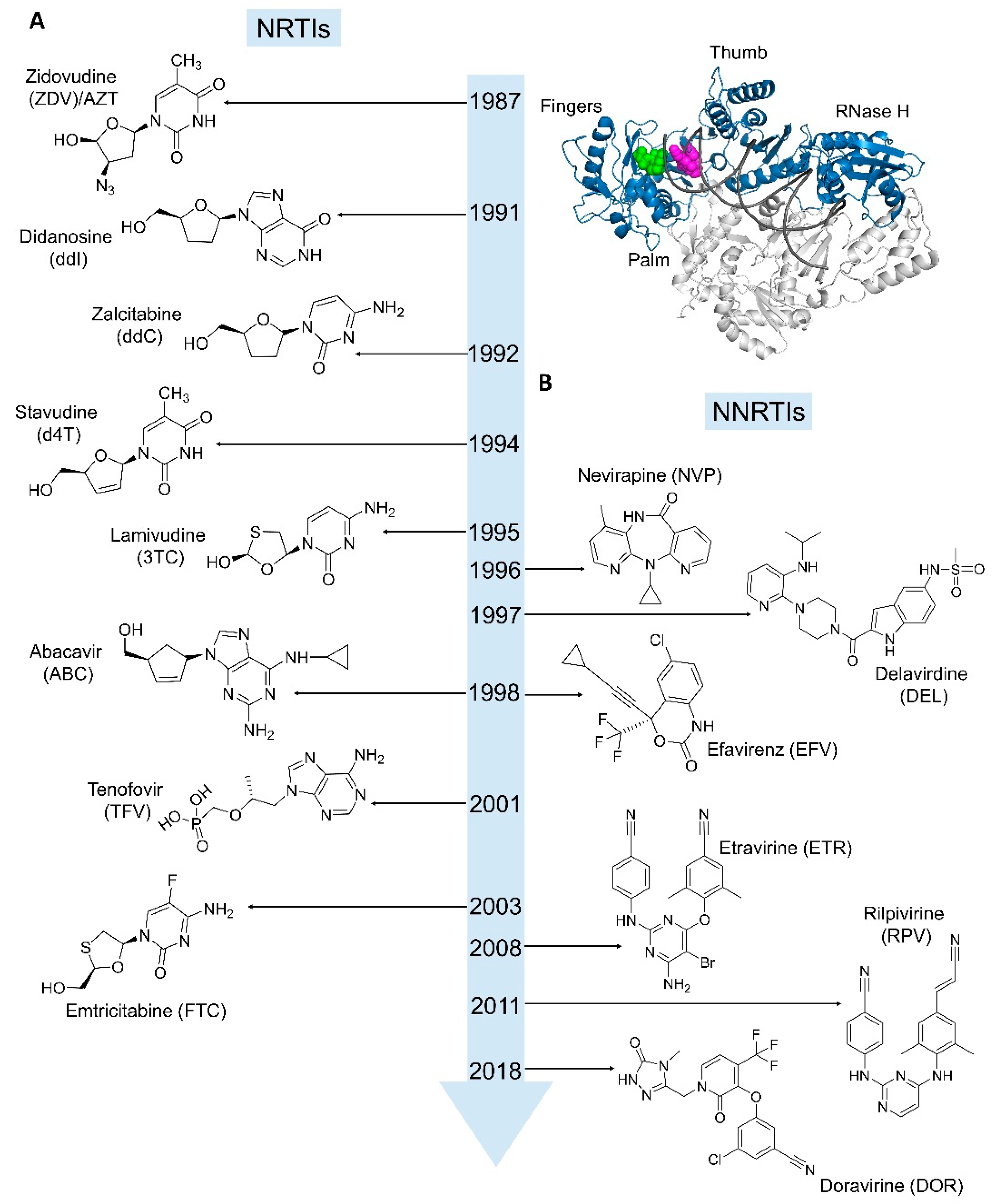
4.1. Nucleoside/Nucleotide RT Inhibitors (NRTIs)
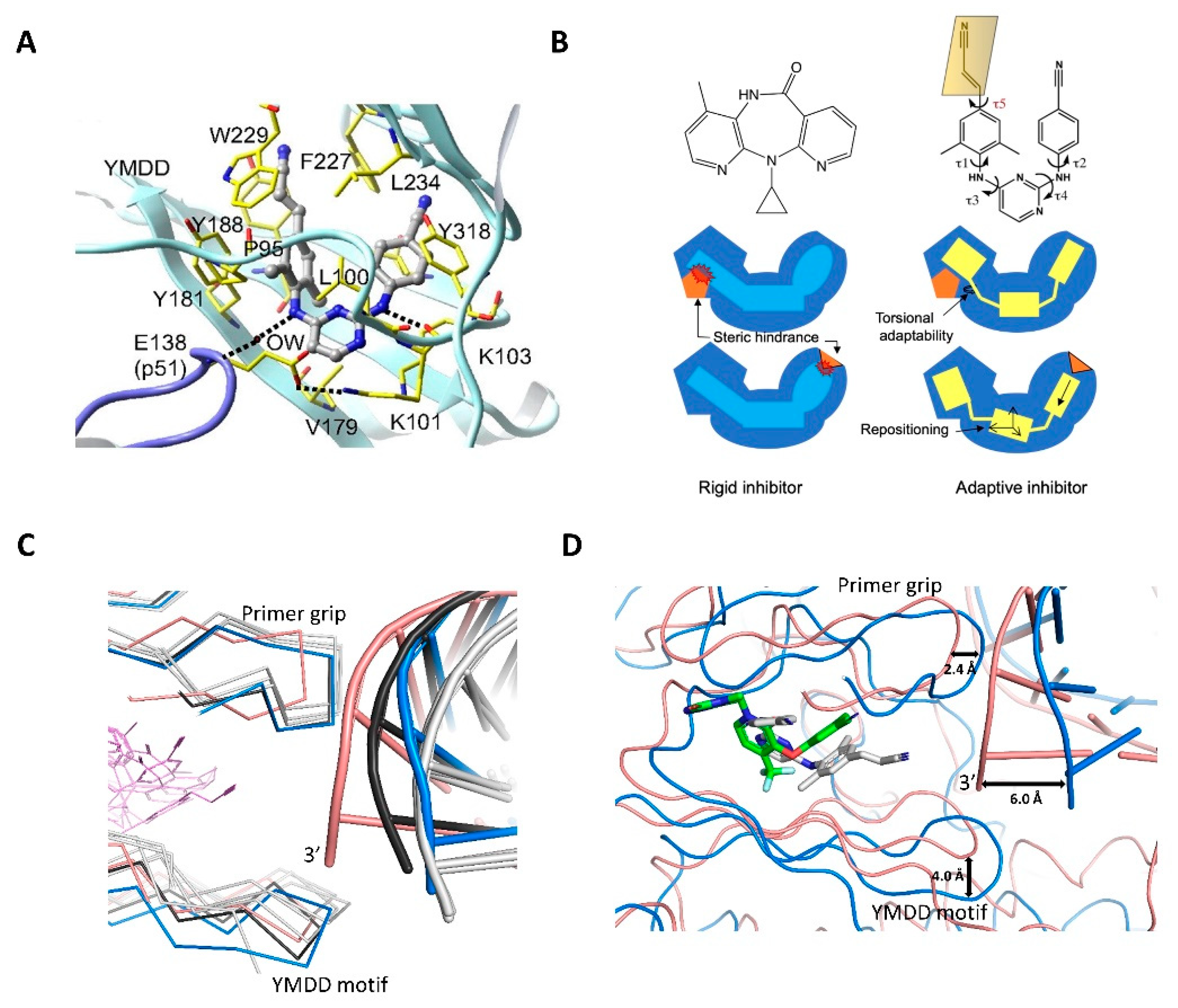
4.2. Non-Nucleoside RT Inhibitors (NNRTIs)
5. Other Targets of RT and Future Perspective
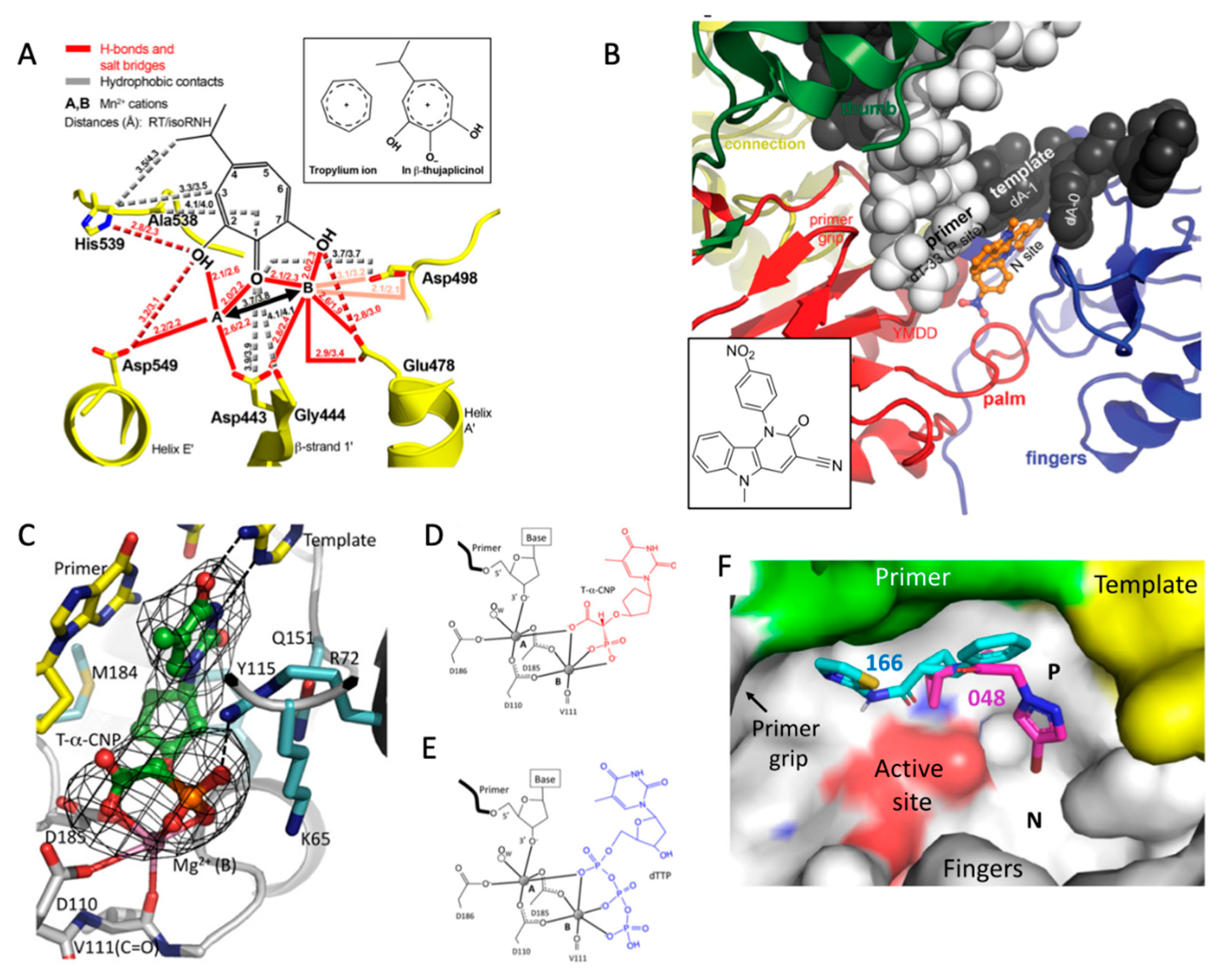
5.1. RNase H
5.2. Nucleoside-Competing RT Inhibitors (NcRTIs)
5.3. P-Pocket
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Temin, H.M.; Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, S.; Boettiger, D.; Temin, H.M. A DNA-depenent DNA polymerase and a DNA endonuclease in virions of Rous sarcoma virus. Nature 1970, 228, 424–427. [Google Scholar] [CrossRef]
- Gilboa, E.; Mitra, S.W.; Goff, S.; Baltimore, D. A detailed model of reverse transcription and tests of crucial aspects. Cell 1979, 18, 93–100. [Google Scholar] [CrossRef]
- Krug, M.S.; Berger, S.L. Ribonuclease H activities associated with viral reverse transcriptases are endonucleases. Proc. Natl. Acad. Sci. USA 1989, 86, 3539–3543. [Google Scholar] [CrossRef] [Green Version]
- MMWR Weekly. 1981. Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/june_5.htm (accessed on 1 March 2022).
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [Green Version]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Vogt, V.M. Retroviral Virions and Genomes. In Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Ratner, L.; Haseltine, W.; Patarca, R.; Livak, K.J.; Starcich, B.; Josephs, S.F.; Doran, E.R.; Rafalski, J.A.; Whitehorn, E.A.; Baumeister, K.; et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 1985, 313, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzon, T.; Leschonsky, B.; Bieler, K.; Paulus, C.; Schroder, J.; Wolf, H.; Wagner, R. Proline residues in the HIV-1 NH2-terminal capsid domain: Structure determinants for proper core assembly and subsequent steps of early replication. Virology 2000, 268, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [Green Version]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Katz, R.A.; Skalka, A.M. The retroviral enzymes. Annu. Rev. Biochem. 1994, 63, 133–173. [Google Scholar] [CrossRef]
- Peliska, J.A.; Benkovic, S.J. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science 1992, 258, 1112–1118. [Google Scholar] [CrossRef]
- Huber, H.E.; Richardson, C.C. Processing of the primer for plus strand DNA synthesis by human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 1990, 265, 10565–10573. [Google Scholar] [CrossRef]
- Rausch, J.W.; Le Grice, S.F. ′Binding, bending and bonding′: Polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. Int. J. Biochem. Cell Biol. 2004, 36, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Basu, V.P.; Song, M.; Gao, L.; Rigby, S.T.; Hanson, M.N.; Bambara, R.A. Strand transfer events during HIV-1 reverse transcription. Virus Res. 2008, 134, 19–38. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. The Interactions of Retroviruses and their Hosts. In Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Le Grice, S.F.; Naas, T.; Wohlgensinger, B.; Schatz, O. Subunit-selective mutagenesis indicates minimal polymerase activity in heterodimer-associated p51 HIV-1 reverse transcriptase. EMBO J. 1991, 10, 3905–3911. [Google Scholar] [CrossRef]
- Di Marzo Veronese, F.; Copeland, T.D.; DeVico, A.L.; Rahman, R.; Oroszlan, S.; Gallo, R.C.; Sarngadharan, M.G. Characterization of highly immunogenic p66/p51 as the reverse transcriptase of HTLV-III/LAV. Science 1986, 231, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.M.; Aitken, A.; Bradley, C.; Darby, G.K.; Larder, B.A.; Powell, K.L.; Purifoy, D.J.; Tisdale, M.; Stammers, D.K. HIV-1 reverse transcriptase: Crystallization and analysis of domain structure by limited proteolysis. Biochemistry 1988, 27, 8884–8889. [Google Scholar] [CrossRef] [PubMed]
- London, R.E. HIV-1 Reverse Transcriptase: A Metamorphic Protein with Three Stable States. Structure 2019, 27, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Arnold, E.; Jacobo-Molina, A.; Nanni, R.G.; Williams, R.L.; Lu, X.; Ding, J.; Clark, A.D., Jr.; Zhang, A.; Ferris, A.L.; Clark, P.; et al. Structure of HIV-1 reverse transcriptase/DNA complex at 7 A resolution showing active site locations. Nature 1992, 357, 85–89. [Google Scholar] [CrossRef]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D., Jr.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef] [Green Version]
- Steitz, T.A. DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larder, B.A.; Purifoy, D.J.; Powell, K.L.; Darby, G. Site-specific mutagenesis of AIDS virus reverse transcriptase. Nature 1987, 327, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; Le Grice, S.F.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance: Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.L.; Kakuda, T.N.; Lichtenstein, K.A. The cellular pharmacology of nucleoside- and nucleotide-analogue reverse-transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 2004, 38, 743–753. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Michailidis, E.; Marchand, B.; Kodama, E.N.; Singh, K.; Matsuoka, M.; Kirby, K.A.; Ryan, E.M.; Sawani, A.M.; Nagy, E.; Ashida, N.; et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-Ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [Google Scholar] [CrossRef] [Green Version]
- Salie, Z.L.; Kirby, K.A.; Michailidis, E.; Marchand, B.; Singh, K.; Rohan, L.C.; Kodama, E.N.; Mitsuya, H.; Parniak, M.A.; Sarafianos, S.G. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. USA 2016, 113, 9274–9279. [Google Scholar] [CrossRef] [Green Version]
- Deval, J. Antimicrobial strategies: Inhibition of viral polymerases by 3′-hydroxyl nucleosides. Drugs 2009, 69, 151–166. [Google Scholar] [CrossRef]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef]
- McQuaid, T.; Savini, C.; Seyedkazemi, S. Sofosbuvir, a Significant Paradigm Change in HCV Treatment. J. Clin. Transl. Hepatol. 2015, 3, 27–35. [Google Scholar] [PubMed] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Cox, R.M.; Wolf, J.D.; Plemper, R.K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. [Google Scholar] [CrossRef]
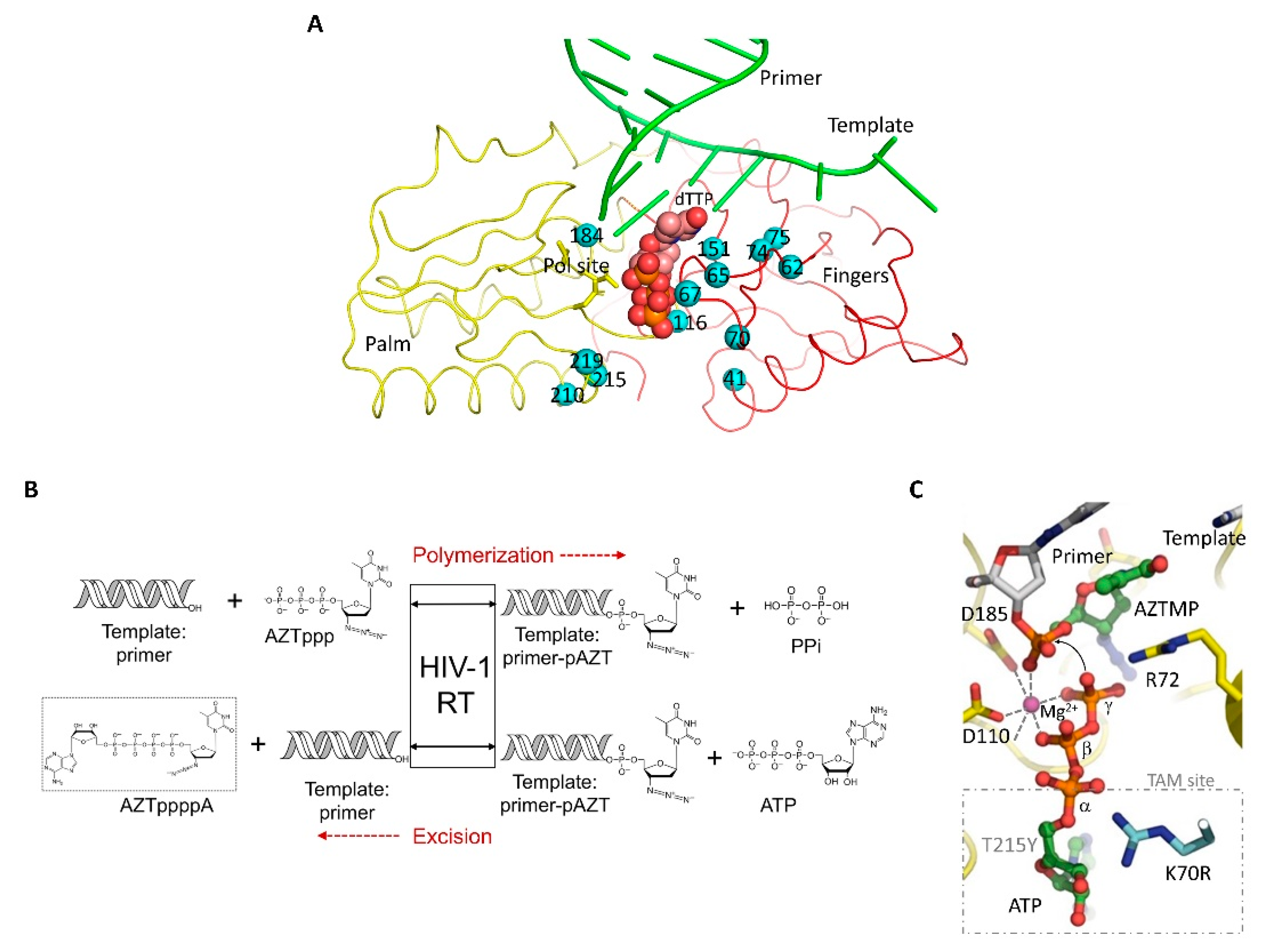
- Menéndez-Arias, L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008, 134, 124–146. [Google Scholar] [CrossRef]
- Deval, J.; Courcambeck, J.; Selmi, B.; Boretto, J.; Canard, B. Structural determinants and molecular mechanisms for the resistance of HIV-1 RT to nucleoside analogues. Curr. Drug Metab. 2004, 5, 305–316. [Google Scholar] [CrossRef]
- Goldschmidt, V.; Marquet, R. Primer unblocking by HIV-1 reverse transcriptase and resistance to nucleoside RT inhibitors (NRTIs). Int. J. Biochem. Cell Biol. 2004, 36, 1687–1705. [Google Scholar] [CrossRef]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance: Part 2. Curr. Opin. Virol. 2013, 3, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Shafer, R.W.; Rhee, S.Y.; Pillay, D.; Miller, V.; Sandstrom, P.; Schapiro, J.M.; Kuritzkes, D.R.; Bennett, D. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS 2007, 21, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Larder, B.A.; Kemp, S.D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science 1989, 246, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Kellam, P.; Boucher, C.A.; Larder, B.A. Fifth mutation in human immunodeficiency virus type 1 reverse transcriptase contributes to the development of high-level resistance to zidovudine. Proc. Natl. Acad. Sci. USA 1992, 89, 1934–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larder, B.A. Interactions between drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase. J. Gen. Virol. 1994, 75, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Kaushik, N.; McCormick, S.; Borkow, G.; Parniak, M.A. Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): Increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 1998, 37, 15908–15917. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; So, A.G.; Scott, W.A. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA 1998, 95, 13471–13476. [Google Scholar] [CrossRef] [Green Version]
- Arion, D.; Parniak, M.A. HIV resistance to zidovudine: The role of pyrophosphorolysis. Drug Resist. Updat. 1999, 2, 91–95. [Google Scholar] [CrossRef]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 2001, 75, 4832–4842. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Das, K.; Han, Q.; Bauman, J.D.; Clark, A.D., Jr.; Hou, X.; Frenkel, Y.V.; Gaffney, B.L.; Jones, R.A.; Boyer, P.L.; et al. Structural basis of HIV-1 resistance to AZT by excision. Nat. Struct. Mol. Biol. 2010, 17, 1202–1209. [Google Scholar] [CrossRef] [Green Version]
- Scott, W.A. Structures of reverse transcriptase pre- and post-excision complexes shed new light on HIV-1 AZT resistance. Viruses 2011, 3, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Winters, M.A.; Coolley, K.L.; Girard, Y.A.; Levee, D.J.; Hamdan, H.; Shafer, R.W.; Katzenstein, D.A.; Merigan, T.C. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J. Clin. Investig. 1998, 102, 1769–1775. [Google Scholar] [CrossRef] [Green Version]
- De Jong, J.J.; Goudsmit, J.; Lukashov, V.V.; Hillebrand, M.E.; Baan, E.; Huismans, R.; Danner, S.A.; ten Veen, J.H.; de Wolf, F.; Jurriaans, S. Insertion of two amino acids combined with changes in reverse transcriptase containing tyrosine-215 of HIV-1 resistant to multiple nucleoside analogs. AIDS 1999, 13, 75–80. [Google Scholar] [CrossRef]
- White, K.L.; Chen, J.M.; Margot, N.A.; Wrin, T.; Petropoulos, C.J.; Naeger, L.K.; Swaminathan, S.; Miller, M.D. Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob. Agents Chemother. 2004, 48, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Sluis-Cremer, N.; Wainberg, M.A.; Schinazi, R.F. Resistance to reverse transcriptase inhibitors used in the treatment and prevention of HIV-1 infection. Future Microbiol. 2015, 10, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- Winters, M.A.; Shafer, R.W.; Jellinger, R.A.; Mamtora, G.; Gingeras, T.; Merigan, T.C. Human immunodeficiency virus type 1 reverse transcriptase genotype and drug susceptibility changes in infected individuals receiving dideoxyinosine monotherapy for 1 to 2 years. Antimicrob. Agents Chemother. 1997, 41, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Margot, N.A.; Lu, B.; Cheng, A.; Miller, M.D. Resistance development over 144 weeks in treatment-naive patients receiving tenofovir disoproxil fumarate or stavudine with lamivudine and efavirenz in Study 903. HIV Med. 2006, 7, 442–450. [Google Scholar] [CrossRef]
- Harrigan, P.R.; Stone, C.; Griffin, P.; Najera, I.; Bloor, S.; Kemp, S.; Tisdale, M.; Larder, B. Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. CNA2001 Investigative Group. J. Infect. Dis. 2000, 181, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Sluis-Cremer, N.; Arion, D.; Kaushik, N.; Lim, H.; Parniak, M.A. Mutational analysis of Lys65 of HIV-1 reverse transcriptase. Biochem. J. 2000, 348 Pt 1, 77–82. [Google Scholar] [CrossRef]
- Das, K.; Bandwar, R.P.; White, K.L.; Feng, J.Y.; Sarafianos, S.G.; Tuske, S.; Tu, X.; Clark, A.D., Jr.; Boyer, P.L.; Hou, X.; et al. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J. Biol. Chem. 2009, 284, 35092–35100. [Google Scholar] [CrossRef] [Green Version]
- Sarafianos, S.G.; Das, K.; Clark, A.D., Jr.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with b-branched amino acids. Proc. Natl. Acad. Sci. USA 1999, 96, 10027–10032. [Google Scholar] [CrossRef] [Green Version]
- Hung, M.; Tokarsky, E.J.; Lagpacan, L.; Zhang, L.; Suo, Z.; Lansdon, E.B. Elucidating molecular interactions of L-nucleotides with HIV-1 reverse transcriptase and mechanism of M184V-caused drug resistance. Commun. Biol. 2019, 2, 469. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar] [CrossRef] [Green Version]
- Yasutake, Y.; Hattori, S.I.; Tamura, N.; Matsuda, K.; Kohgo, S.; Maeda, K.; Mitsuya, H. Structural features in common of HBV and HIV-1 resistance against chirally-distinct nucleoside analogues entecavir and lamivudine. Sci. Rep. 2020, 10, 3021. [Google Scholar] [CrossRef] [Green Version]
- Jilek, B.L.; Zarr, M.; Sampah, M.E.; Rabi, S.A.; Bullen, C.K.; Lai, J.; Shen, L.; Siliciano, R.F. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 2012, 18, 446–451. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Y.; Ly, J.K.; Myrick, F.; Goodman, D.; White, K.L.; Svarovskaia, E.S.; Borroto-Esoda, K.; Miller, M.D. The triple combination of tenofovir, emtricitabine and efavirenz shows synergistic anti-HIV-1 activity in vitro: A mechanism of action study. Retrovirology 2009, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.T.; Hitchcock, M.J. Cellular pharmacology of 2′,3′-dideoxy-2′,3′-didehydrothymidine, a nucleoside analog active against human immunodeficiency virus. Antimicrob. Agents Chemother. 1989, 33, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Bethell, R.C.; Lie, Y.S.; Parkin, N.T. In vitro activity of SPD754, a new deoxycytidine nucleoside reverse transcriptase inhibitor (NRTI), against 215 HIV-1 isolates resistant to other NRTIs. Antivir. Chem. Chemother. 2005, 16, 295–302. [Google Scholar] [CrossRef] [Green Version]
- De Baar, M.P.; de Rooij, E.R.; Smolders, K.G.; van Schijndel, H.B.; Timmermans, E.C.; Bethell, R. Effects of apricitabine and other nucleoside reverse transcriptase inhibitors on replication of mitochondrial DNA in HepG2 cells. Antivir. Res. 2007, 76, 68–74. [Google Scholar] [CrossRef]
- Lin, T.S.; Luo, M.Z.; Liu, M.C.; Zhu, Y.L.; Gullen, E.; Dutschman, G.E.; Cheng, Y.C. Design and synthesis of 2′,3′-dideoxy-2′,3′-didehydro-beta-L-cytidine (beta-L-d4C) and 2′,3′-dideoxy 2′,3′-didehydro-beta-L-5-fluorocytidine (beta-L-Fd4C), two exceptionally potent inhibitors of human hepatitis B virus (HBV) and potent inhibitors of human immunodeficiency virus (HIV) in vitro. J. Med. Chem. 1996, 39, 1757–1759. [Google Scholar]
- Michailidis, E.; Huber, A.D.; Ryan, E.M.; Ong, Y.T.; Leslie, M.D.; Matzek, K.B.; Singh, K.; Marchand, B.; Hagedorn, A.N.; Kirby, K.A.; et al. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J. Biol. Chem. 2014, 289, 24533–24548. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, A.; Kodama, E.; Sarafianos, S.G.; Sakagami, Y.; Kohgo, S.; Kitano, K.; Ashida, N.; Iwai, Y.; Hayakawa, H.; Nakata, H.; et al. 2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. [Google Scholar] [CrossRef]
- Kirby, K.A.; Michailidis, E.; Fetterly, T.L.; Steinbach, M.A.; Singh, K.; Marchand, B.; Leslie, M.D.; Hagedorn, A.N.; Kodama, E.N.; Marquez, V.E.; et al. Effects of substitutions at the 4′ and 2 positions on the bioactivity of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 2013, 57, 6254–6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, E.I.; Kohgo, S.; Kitano, K.; Machida, H.; Gatanaga, H.; Shigeta, S.; Matsuoka, M.; Ohrui, H.; Mitsuya, H. 4′-Ethynyl nucleoside analogs: Potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 2001, 45, 1539–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, H.; Kohgo, S.; Kitano, K.; Ashida, N.; Kodama, E.; Mitsuya, H.; Ohrui, H. Potential of 4′-C-substituted nucleosides for the treatment of HIV-1. Antivir. Chem. Chemother. 2004, 15, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Sohl, C.D.; Singh, K.; Kasiviswanathan, R.; Copeland, W.C.; Mitsuya, H.; Sarafianos, S.G.; Anderson, K.S. Mechanism of interaction of human mitochondrial DNA polymerase gamma with the novel nucleoside reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine indicates a low potential for host toxicity. Antimicrob. Agents Chemother. 2012, 56, 1630–1634. [Google Scholar] [CrossRef] [Green Version]
- Merluzzi, V.J.; Hargrave, K.D.; Labadia, M.; Grozinger, K.; Skoog, M.; Wu, J.C.; Shih, C.K.; Eckner, K.; Hattox, S.; Adams, J.; et al. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science 1990, 250, 1411–1413. [Google Scholar] [CrossRef]
- Pauwels, R.; Andries, K.; Desmyter, J.; Schols, D.; Kukla, M.J.; Breslin, H.J.; Raeymaeckers, A.; Van Gelder, J.; Woestenborghs, R.; Heykants, J.; et al. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 1990, 343, 470–474. [Google Scholar] [CrossRef]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark, A.D., Jr.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef]
- Hsiou, Y.; Ding, J.; Das, K.; Clark, A.D., Jr.; Hughes, S.H.; Arnold, E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: Implications of conformational changes for polymerization and inhibition mechanisms. Structure 1996, 4, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Richman, D.D.; Havlir, D.; Corbeil, J.; Looney, D.; Ignacio, C.; Spector, S.A.; Sullivan, J.; Cheeseman, S.; Barringer, K.; Pauletti, D.; et al. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J. Virol. 1994, 68, 1660–1666. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.B.; Becker-Pergola, G.; Guay, L.A.; Musoke, P.; Mracna, M.; Fowler, M.G.; Mofenson, L.M.; Mirochnick, M.; Mmiro, F.; Eshleman, S.H. Identification of the K103N resistance mutation in Ugandan women receiving nevirapine to prevent HIV-1 vertical transmission. AIDS 2000, 14, F111–F115. [Google Scholar] [CrossRef]
- Cunningham, C.K.; Chaix, M.L.; Rekacewicz, C.; Britto, P.; Rouzioux, C.; Gelber, R.D.; Dorenbaum, A.; Delfraissy, J.F.; Bazin, B.; Mofenson, L.; et al. Development of resistance mutations in women receiving standard antiretroviral therapy who received intrapartum nevirapine to prevent perinatal human immunodeficiency virus type 1 transmission: A substudy of pediatric AIDS clinical trials group protocol 316. J. Infect. Dis 2002, 186, 181–188. [Google Scholar] [PubMed]
- Singh, K.; Marchand, B.; Rai, D.K.; Sharma, B.; Michailidis, E.; Ryan, E.M.; Matzek, K.B.; Leslie, M.D.; Hagedorn, A.N.; Li, Z.; et al. Biochemical Mechanism of HIV-1 Resistance to Rilpivirine. J. Biol. Chem. 2012, 287, 38110–38123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maga, G.; Amacker, M.; Ruel, N.; Hubscher, U.; Spadari, S. Resistance to nevirapine of HIV-1 reverse transcriptase mutants: Loss of stabilizing interactions and thermodynamic or steric barriers are induced by different single amino acid substitutions. J. Mol. Biol. 1997, 274, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Ding, J.; Hsiou, Y.; Clark, A.D., Jr.; Moereels, H.; Koymans, L.; Andries, K.; Pauwels, R.; Janssen, P.A.; Boyer, P.L.; et al. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 1996, 264, 1085–1100. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef]
- Ding, J.; Das, K.; Moereels, H.; Koymans, L.; Andries, K.; Janssen, P.A.; Hughes, S.H.; Arnold, E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 407–415. [Google Scholar] [CrossRef]
- Ren, J.; Milton, J.; Weaver, K.L.; Short, S.A.; Stuart, D.I.; Stammers, D.K. Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure 2000, 8, 1089–1094. [Google Scholar] [CrossRef]
- Lindberg, J.; Sigurdsson, S.; Lowgren, S.; Andersson, H.O.; Sahlberg, C.; Noreen, R.; Fridborg, K.; Zhang, H.; Unge, T. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 2002, 269, 1670–1677. [Google Scholar] [CrossRef]
- Janssen, P.A.; Lewi, P.J.; Arnold, E.; Daeyaert, F.; de Jonge, M.; Heeres, J.; Koymans, L.; Vinkers, M.; Guillemont, J.; Pasquier, E.; et al. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J. Med. Chem. 2005, 48, 1901–1909. [Google Scholar] [CrossRef]
- Das, K.; Bauman, J.D.; Clark, A.D., Jr.; Frenkel, Y.V.; Lewi, P.J.; Shatkin, A.J.; Hughes, S.H.; Arnold, E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. USA 2008, 105, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Clark, J.A.D.; Lewi, P.J.; Heeres, J.; de Jonge, M.R.; Koymans, L.M.H.; Vinkers, H.M.; Daeyaert, F.; Ludovici, D.W.; Kukla, M.J.; et al. Roles of Conformational and Positional Adaptability in Structure-Based Design of TMC125-R165335 (Etravirine) and Related Non-nucleoside Reverse Transcriptase Inhibitors That Are Highly Potent and Effective against Wild-Type and Drug-Resistant HIV-1 Variants. J. Med. Chem. 2004, 47, 2550–2560. [Google Scholar] [PubMed]
- Smith, S.J.; Pauly, G.T.; Akram, A.; Melody, K.; Ambrose, Z.; Schneider, J.P.; Hughes, S.H. Rilpivirine and Doravirine Have Complementary Efficacies Against NNRTI-Resistant HIV-1 Mutants. J. Acquir. Immune Defic. Syndr. 2016, 72, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, K.P.; Mathiharan, Y.K.; Kappel, K.; Coey, A.T.; Chen, D.H.; Barrero, D.; Madigan, L.; Puglisi, J.D.; Skiniotis, G.; Puglisi, E.V. Architecture of an HIV-1 reverse transcriptase initiation complex. Nature 2018, 557, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Martinez, S.E.; DeStefano, J.J.; Arnold, E. Structure of HIV-1 RT/dsRNA initiation complex prior to nucleotide incorporation. Proc. Natl. Acad. Sci. USA 2019, 116, 7308–7313. [Google Scholar] [CrossRef] [Green Version]
- Lanchy, J.M.; Ehresmann, C.; Le Grice, S.F.; Ehresmann, B.; Marquet, R. Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J. 1996, 15, 7178–7187. [Google Scholar] [CrossRef]
- Ha, B.; Larsen, K.P.; Zhang, J.; Fu, Z.; Montabana, E.; Jackson, L.N.; Chen, D.H.; Puglisi, E.V. High-resolution view of HIV-1 reverse transcriptase initiation complexes and inhibition by NNRTI drugs. Nat. Commun. 2021, 12, 2500. [Google Scholar] [CrossRef]
- Singh, A.K.; De Wijngaert, B.; Bijnens, M.; Uyttersprot, K.; Nguyen, H.; Martinez, S.E.; Schols, D.; Herdewijn, P.; Pannecouque, C.; Arnold, E.; et al. Cryo-EM structures of Doravirine and Rilpivirine with HIV-1 Reverse Transcriptase/DNA Aptamer–Nonnucleoside Inhibitor Resistance by E138K and M184I Mutations. bioRxiv 2022. [Google Scholar] [CrossRef]
- Davies, J.F., II; Hostomska, Z.; Hostomsky, Z.; Jordan, S.R.; Matthews, D.A. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 1991, 252, 88–95. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Das, K.; Tantillo, C.; Clark, A.D., Jr.; Ding, J.; Whitcomb, J.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001, 20, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Lapkouski, M.; Tian, L.; Miller, J.T.; Le Grice, S.F.; Yang, W. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat. Struct. Mol. Biol. 2013, 20, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Martinez, S.E.; Bandwar, R.P.; Arnold, E. Structures of HIV-1 RT-RNA/DNA ternary complexes with dATP and nevirapine reveal conformational flexibility of RNA/DNA: Insights into requirements for RNase H cleavage. Nucleic Acids Res. 2014, 42, 8125–8137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Kim, M.S.; Li, H.; Wang, J.; Yang, W. Structure of HIV-1 reverse transcriptase cleaving RNA in an RNA/DNA hybrid. Proc. Natl. Acad. Sci. USA 2018, 115, 507–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grobler, J.A.; Stillmock, K.; Hu, B.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J.; et al. Diketo acid inhibitor mechanism and HIV-1 integrase: Implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 6661–6666. [Google Scholar] [CrossRef] [Green Version]
- Klumpp, K.; Hang, J.Q.; Rajendran, S.; Yang, Y.; Derosier, A.; Wong Kai In, P.; Overton, H.; Parkes, K.E.; Cammack, N.; Martin, J.A. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003, 31, 6852–6859. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef]
- Shaw-Reid, C.A.; Munshi, V.; Graham, P.; Wolfe, A.; Witmer, M.; Danzeisen, R.; Olsen, D.B.; Carroll, S.S.; Embrey, M.; Wai, J.S.; et al. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 2003, 278, 2777–2780. [Google Scholar] [CrossRef] [Green Version]
- Himmel, D.M.; Maegley, K.A.; Pauly, T.A.; Bauman, J.D.; Das, K.; Dharia, C.; Clark, A.D., Jr.; Ryan, K.; Hickey, M.J.; Love, R.A.; et al. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure 2009, 17, 1625–1635. [Google Scholar] [CrossRef] [Green Version]
- Su, H.P.; Yan, Y.; Prasad, G.S.; Smith, R.F.; Daniels, C.L.; Abeywickrema, P.D.; Reid, J.C.; Loughran, H.M.; Kornienko, M.; Sharma, S.; et al. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 2010, 84, 7625–7633. [Google Scholar] [CrossRef] [Green Version]
- Lansdon, E.B.; Liu, Q.; Leavitt, S.A.; Balakrishnan, M.; Perry, J.K.; Lancaster-Moyer, C.; Kutty, N.; Liu, X.; Squires, N.H.; Watkins, W.J.; et al. Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxy quinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 2011, 55, 2905–2915. [Google Scholar] [CrossRef] [Green Version]
- Jochmans, D.; Deval, J.; Kesteleyn, B.; Van Marck, H.; Bettens, E.; De Baere, I.; Dehertogh, P.; Ivens, T.; Van Ginderen, M.; Van Schoubroeck, B.; et al. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J. Virol. 2006, 80, 12283–12292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehteshami, M.; Scarth, B.J.; Tchesnokov, E.P.; Dash, C.; Le Grice, S.F.; Hallenberger, S.; Jochmans, D.; Gotte, M. Mutations M184V and Y115F in HIV-1 reverse transcriptase discriminate against “nucleotide-competing reverse transcriptase inhibitors”. J. Biol. Chem. 2008, 283, 29904–29911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, F.X.; Hoang, A.; Das, K.; Arnold, E. Structural Basis of HIV-1 Inhibition by Nucleotide-Competing Reverse Transcriptase Inhibitor INDOPY-1. J. Med. Chem. 2019, 62, 9996–10002. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Das, K.; Bernatchez, J.A.; Martinez, S.E.; Ngure, M.; Keane, S.; Ford, A.; Maguire, N.; Mullins, N.; John, J.; et al. Alpha-carboxy nucleoside phosphonates as universal nucleoside triphosphate mimics. Proc. Natl. Acad. Sci. USA 2015, 112, 3475–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, V.L. Enzymatic transition states and transition state analog design. Annu. Rev. Biochem. 1998, 67, 693–720. [Google Scholar] [CrossRef] [Green Version]
- Schramm, V.L. Enzymatic Transition States and Drug Design. Chem. Rev. 2018, 118, 11194–11258. [Google Scholar] [CrossRef]
- Metzger, W.; Hermann, T.; Schatz, O.; Le Grice, S.F.; Heumann, H. Hydroxyl radical footprint analysis of human immunodeficiency virus reverse transcriptase-template.primer complexes. Proc. Natl. Acad. Sci. USA 1993, 90, 5909–5913. [Google Scholar] [CrossRef] [Green Version]
- DeStefano, J.J.; Mallaber, L.M.; Fay, P.J.; Bambara, R.A. Determinants of the RNase H cleavage specificity of human immunodeficiency virus reverse transcriptase. Nucleic Acids Res. 1993, 21, 4330–4338. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, M.; Balakrishnan, M.; Palaniappan, C.; Fay, P.J.; Bambara, R.A. The sequential mechanism of HIV reverse transcriptase RNase H. J. Biol. Chem. 2000, 275, 37664–37671. [Google Scholar] [CrossRef] [Green Version]
- Winshell, J.; Paulson, B.A.; Buelow, B.D.; Champoux, J.J. Requirements for DNA unpairing during displacement synthesis by HIV-1 reverse transcriptase. J. Biol. Chem. 2004, 279, 52924–52933. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.J.; Berger, S.; Kensch, O.; Felekyan, S.; Antonik, M.; Wohrl, B.M.; Restle, T.; Goody, R.S.; Seidel, C.A. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 1655–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbondanzieri, E.A.; Bokinsky, G.; Rausch, J.W.; Zhang, J.X.; Le Grice, S.F.; Zhuang, X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature 2008, 453, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, L.I.; Stone, M.D. Single-Molecule Analysis of Reverse Transcriptase Enzymes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032458. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Martinez, S.E.; Gu, W.J.; Nguyen, H.; Schols, D.; Herdewijn, P.; De Jonghe, S.; Das, K. Sliding of HIV-1 reverse transcriptase over DNA creates a transient P pocket–targeting P-pocket by fragment screening. Nat. Commun. 2021, 12, 7127. [Google Scholar] [CrossRef] [PubMed]
- Mega, E.R. Alarming surge in drug-resistant HIV uncovered. Nature 2019. Available online: https://europepmc.org/article/med/32724203 (accessed on 5 April 2022).
- Landovitz, R.J.; Donnell, D.; Clement, M.E.; Hanscom, B.; Cottle, L.; Coelho, L.; Cabello, R.; Chariyalertsak, S.; Dunne, E.F.; Frank, I.; et al. Cabotegravir for HIV Prevention in Cisgender Men and Transgender Women. N. Engl. J. Med. 2021, 385, 595–608. [Google Scholar] [CrossRef]
- Canetti, D.; Muccini, C.; Spagnuolo, V.; Galli, L.; Poli, A.; Gianotti, N.; Feasi, M.; Castagna, A. Achieving virological control in pan-resistant HIV-1 infection: A case series. EBioMedicine 2022, 77, 103906. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.K.; Das, K. Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies. Viruses 2022, 14, 1027. https://doi.org/10.3390/v14051027
Singh AK, Das K. Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies. Viruses. 2022; 14(5):1027. https://doi.org/10.3390/v14051027
Chicago/Turabian StyleSingh, Abhimanyu K., and Kalyan Das. 2022. "Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies" Viruses 14, no. 5: 1027. https://doi.org/10.3390/v14051027
APA StyleSingh, A. K., & Das, K. (2022). Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies. Viruses, 14(5), 1027. https://doi.org/10.3390/v14051027