
Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Isolation of the vB_AspA_Bolek, vB_AspA_Lolek, and vB_AspA_Tola Phages
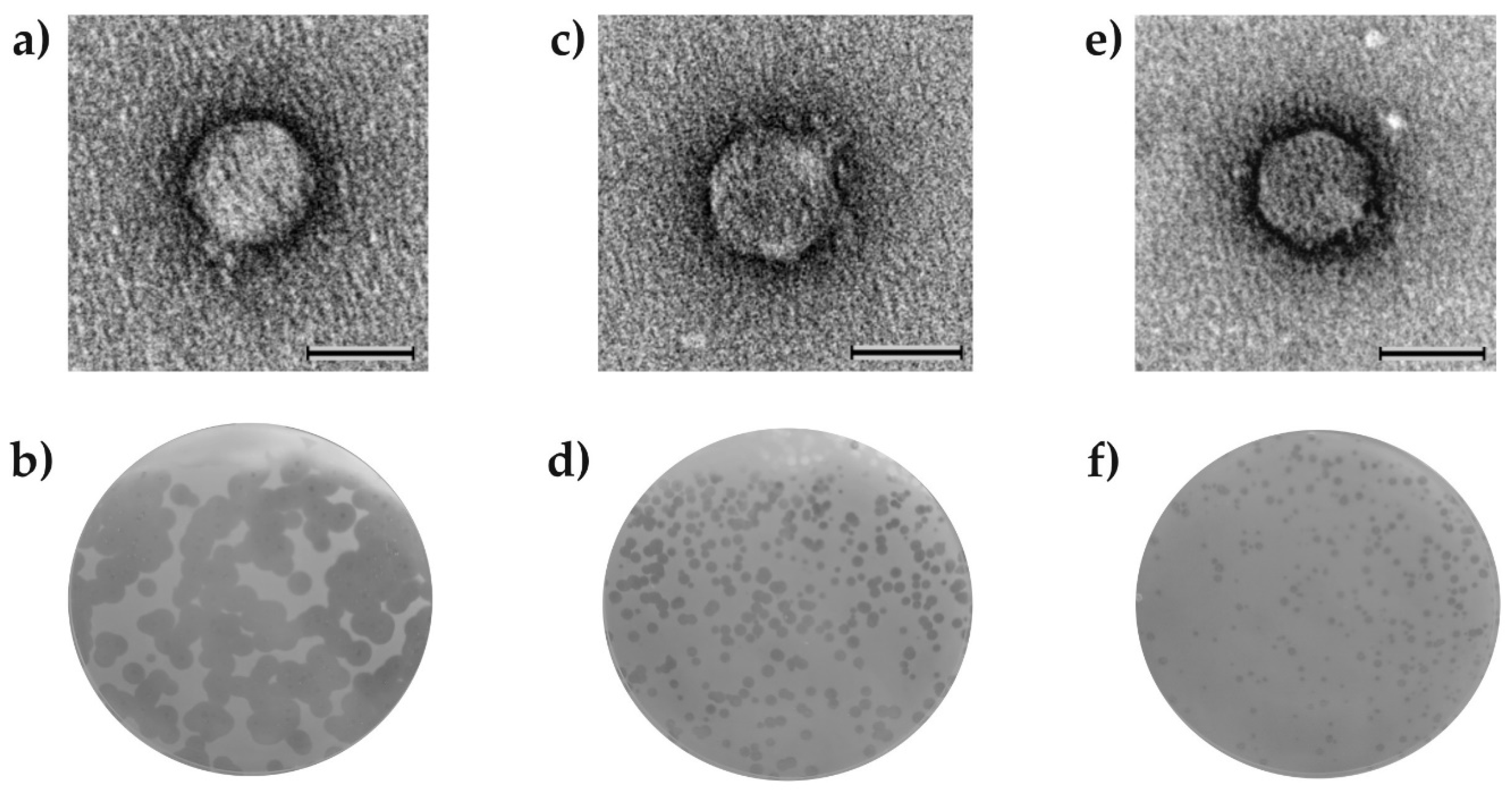
2.3. Transmission Electron Microscopy (TEM)
2.4. Temperature Range of Bacterial Growth
2.5. Bacterial Growth Curve
2.6. Testing the Influence of Various Temperatures on the Phage Plaque Formation
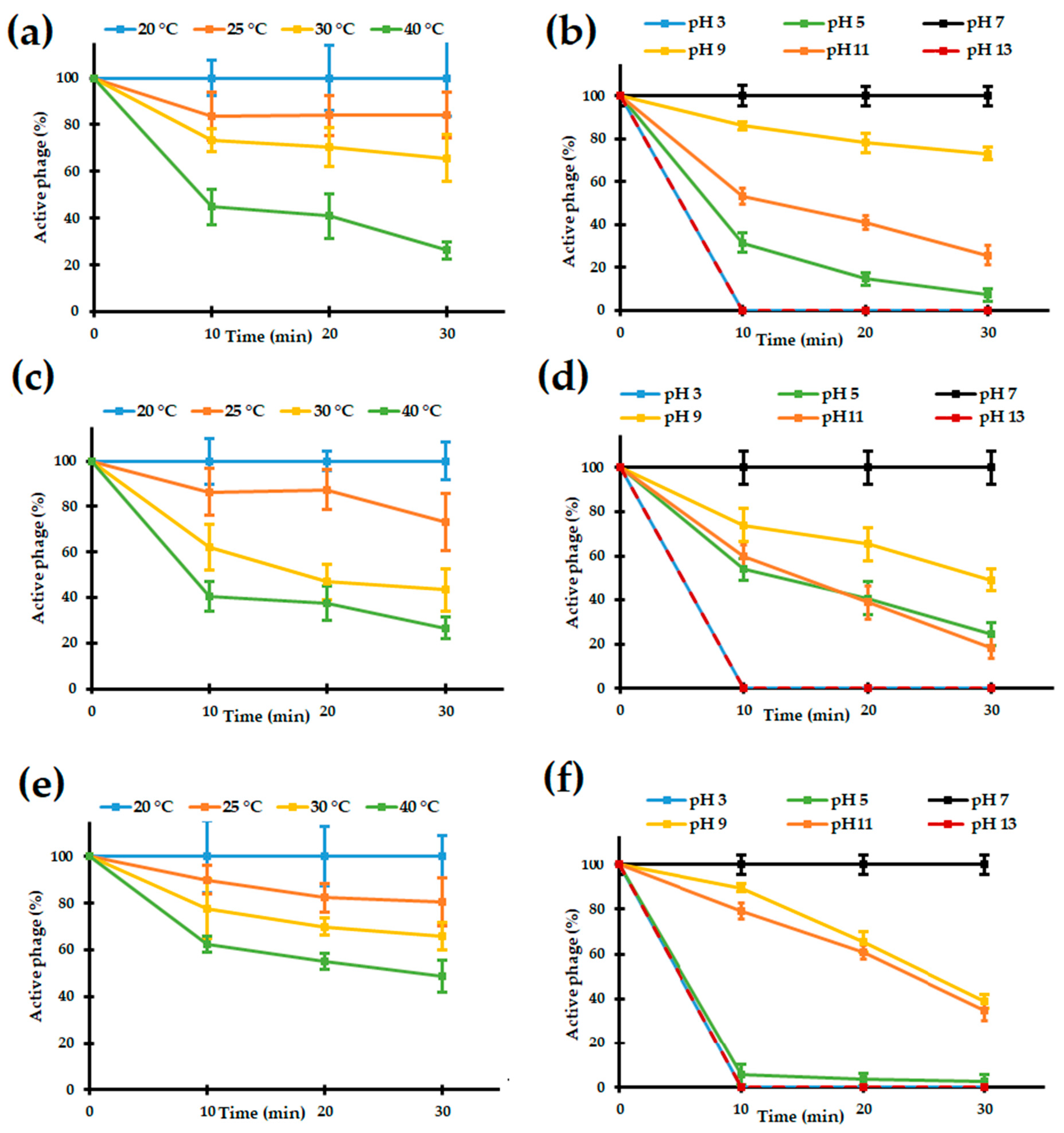
2.7. Thermal and pH Stability of the Phages
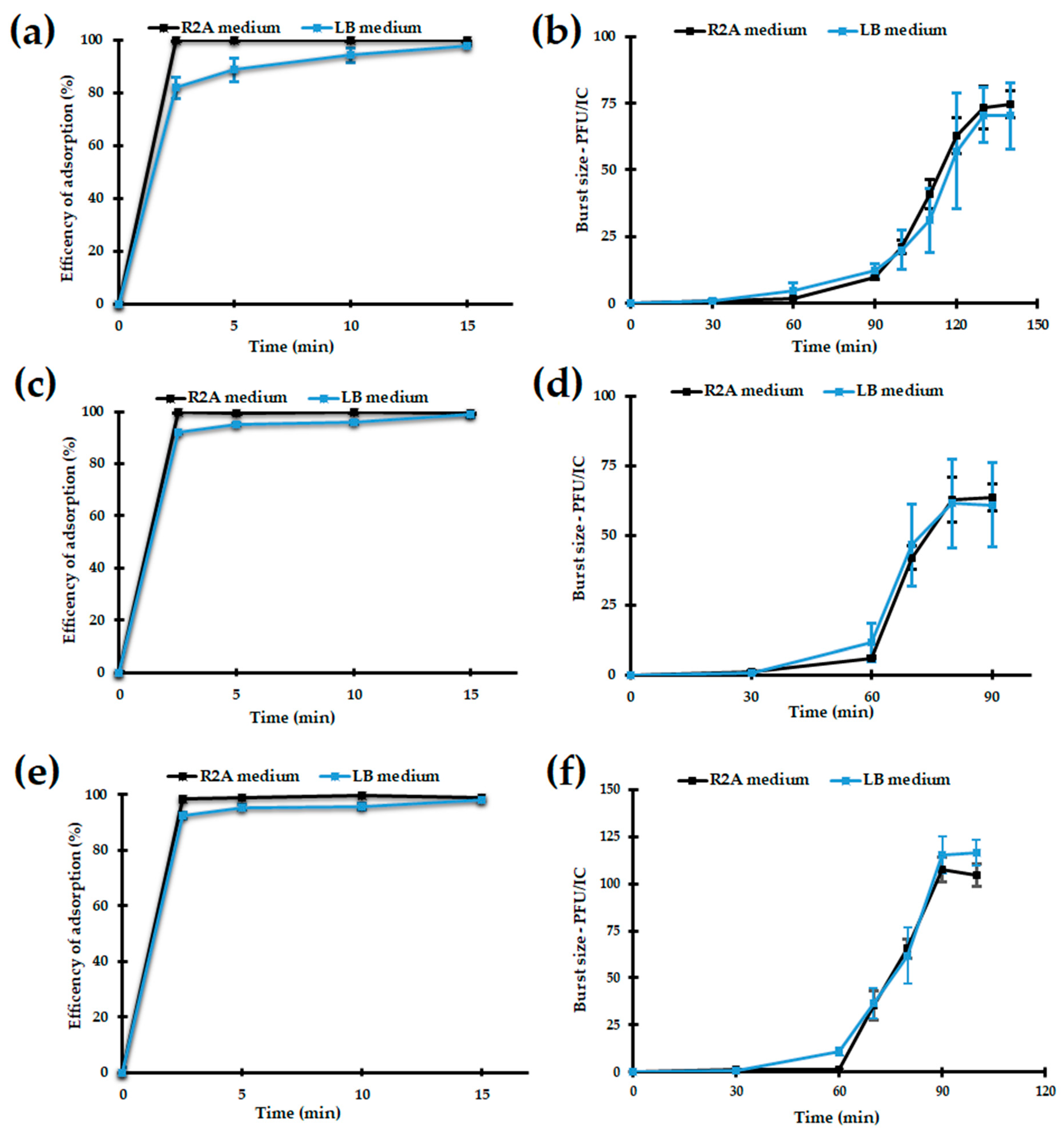
2.8. Adsorption Kinetics
2.9. One-Step Growth Curve
2.10. Determination of Phage Host Range by Spot Testing
2.11. DNA Isolation and Sequencing
2.12. Genome Annotation
2.13. Comparative Genomics Analysis
2.14. Nucleotide Sequence Accession Numbers
3. Results and Discussion
3.1. Identification and characterization of the Hosts
3.2. Identification and Functional characterization of Phages
3.2.1. Identification of the vB_AspA_Bolek, vB_AspA_Lolek, and vB_AspA_Tola Phages
3.2.2. The Influence of Various Temperatures on the Phage Plaque Formation
3.2.3. Sensitivity to Temperature and pH of the Bolek, Lolek, and Tola Phages
3.2.4. Adsorption and One-Step Growth of the Bolek, Lolek, and Tola Phages
3.2.5. Host Range
3.3. Genomic characterization of Phages
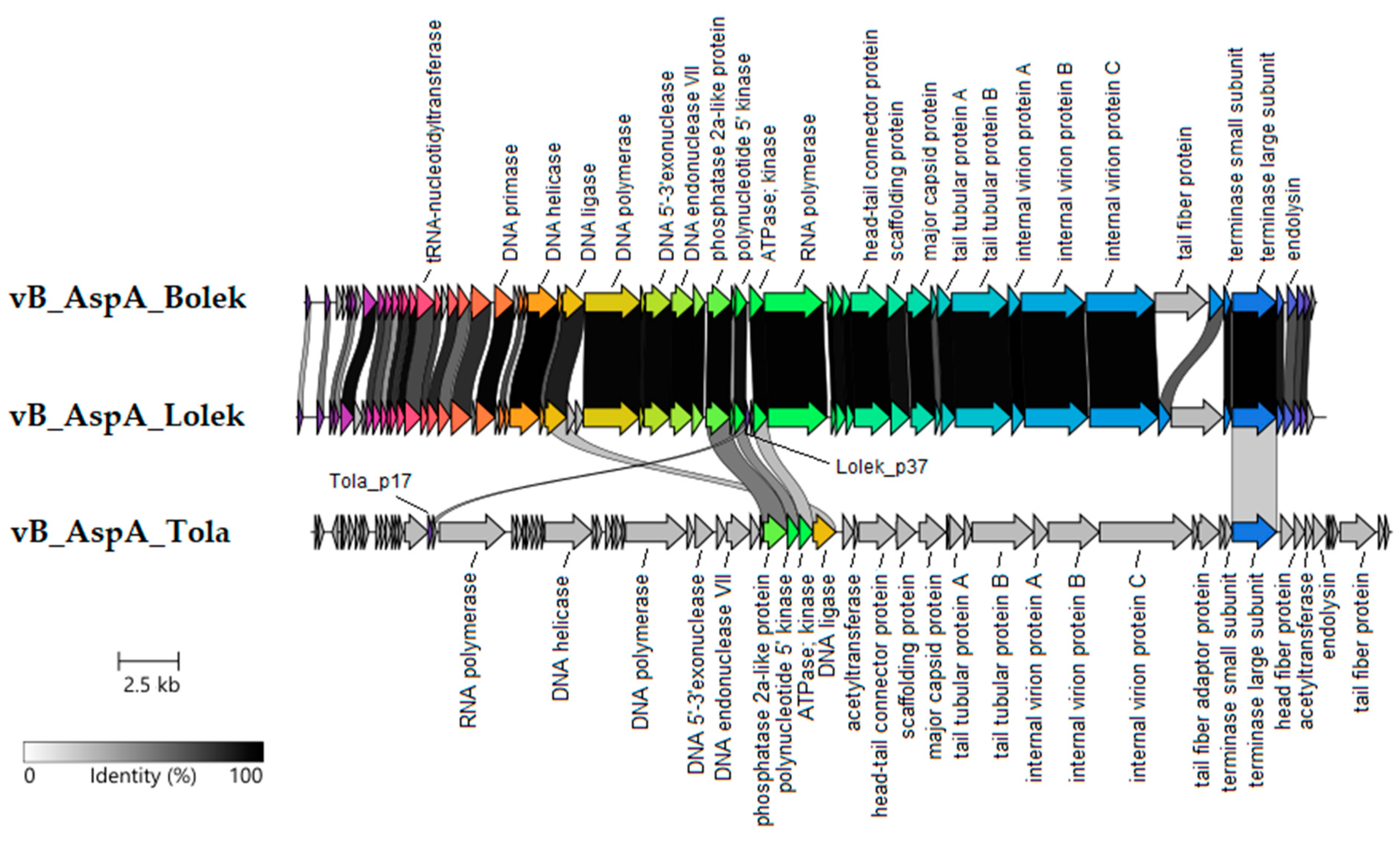
3.4. Comparative Analysis of the Bolek, Lolek, and Tola Phages
3.5. Comparative Analysis of the Bolek, Lolek, and Tola with Other Phages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janda, M.J.; Abbott, S.L. The Genus Aeromonas: Taxonomy, Pathogenicity, and Infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Bravo, A.; Figueras, M.J. An Update on the Genus Aeromonas: Taxonomy, Epidemiology, and Pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyam, B.; Purva, M.; Mahesh, C.M. Aeromonas spp.: An Emerging Nosocomial Pathogen. J. Lab. Physicians 2020, 8, 001–004. [Google Scholar] [CrossRef]
- Dallaire-Dufresne, S.; Tanaka, K.H.; Trudel, M.V.; Lafaille, A.; Charette, S.J. Virulence, genomic features, and plasticity of Aeromonas salmonicida subsp. salmonicida, the causative agent of fish furunculosis. Vet. Microbiol. 2014, 169, 1–7. [Google Scholar] [CrossRef]
- Burr, S.E.; Pugovkin, D.; Wahli, T.; Segner, H.; Frey, J. Attenuated virulence of an Aeromonas salmonicida subsp. salmonicida type III secretion mutant in a rainbow trout model. Microbiology 2005, 151, 2111–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamed, H.; Ibrahim, I.; Baumgartner, W.; Lawrence, M.L.; Karsi, A. Characterization of Histopathological and Ultrastructural Changes in Channel Catfish Experimentally Infected with Virulent Aeromonas hydrophila. Front. Microbiol. 2017, 8, 1519. [Google Scholar] [CrossRef]
- Jin, L.; Chen, Y.; Yang, W.; Qiao, Z.; Zhang, X. Complete genome sequence of fish-pathogenic Aeromonas hydrophila HX-3 and a comparative analysis: Insights into virulence factors and quorum sensing. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Taylor, P.W.; Stapleton, P.D.; Paul, L.J. New ways to treat bacterial infections. Drug Discov. Today 2002, 7, 1086–1091. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis Part 2: Management Strategies and New Agents. Pharm. Ther. 2015, 40, 344–352. [Google Scholar]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage therapy: A potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries. 2014, 8, 129–136. [Google Scholar] [CrossRef]
- Hauser, A.R.; Mecsas, J.; Moir, D.T. Beyond Antibiotics: New Therapeutic Approaches for Bacterial Infections. Clin. Infect. Dis. 2021, 63, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushegian, A.R. Are There 1031 Virus Particles on Earth, or More, or Fewer? J. Bacteriol. 2020, 202, e00052-20. [Google Scholar] [CrossRef] [PubMed]
- Dy, R.L.; Rigano, L.A.; Fineran, P.C. Phage-based biocontrol strategies and their application in agriculture and aquaculture. Biochem. Soc. Trans. 2018, 46, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Akmal, M.; Rahimi-Midani, A.; Hafeez-ur-Rehman, M.; Hussain, A.; Choi, T.J. Isolation, Characterization, and Application of a Bacteriophage Infecting the Fish Pathogen Aeromonas hydrophila. Pathogens 2020, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.W.; Kim, J.H.; Shin, S.P.; Han, J.E.; Chai, J.Y. Protective effects of the Aeromonas phages pAh1-C and pAh6-C against mass mortality of the cyprinid loach (Misgurnus anguillicaudatus) caused by Aeromonas hydrophila. Aquaculture 2021, 416–417, 289–295. [Google Scholar] [CrossRef]
- Kabwe, M.; Brown, T.; Speirs, L.; Kub, H.; Leach, M.; Chan, H.T.; Petrovski, S.; Lock, P.; Tucci, J. Novel Bacteriophages Capable of Disrupting Biofilms From Clinical Strains of Aeromonas hydrophila. Front. Microbiol. 2020, 11, 194. [Google Scholar] [CrossRef]
- Le, T.S.; Nguyen, T.H.; Vo, H.P.; Doan, V.C.; Nguyen, H.L.; Tran, M.T.; Tran, T.T.; Southgate, P.C.; Kurtboke, D.I. Protective Effects of Bacteriophages against Aeromonas hydrophila Causing Motile Aeromonas Septicemia (MAS) in Striped Catfish. Antibiotics 2018, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://talk.ictvonline.org/taxonomy/vmr/m/vmr-file-repository/13181 (accessed on 20 February 2022).
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Kawasaki, T.; Shimizu, M.; Satsuma, H.; Fujiwara, A.; Fujie, M.; Usami, S.; Yamada, T. Genomic Characterization of Ralstonia solanacearum Phage φRSB1, a T7-Like Wide-Host-Range Phage. J. Bacteriol. 2009, 191, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Boeckman, J.; Korn, A.; Yao, G.; Ravindran, A.; Gonzalez, C.; Gillac, J. Sheep in wolves’ clothing: Temperate T7-like bacteriophages and the origins of the Autographiviridae. Virology 2022, 568, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Ely, B.; Berrios, L.; Thomas, Q. S2B, a Temperate Bacteriophage That Infects Caulobacter Crescentus Strain CB15. Curr. Microbiol. 2022, 79, 98. [Google Scholar] [CrossRef] [PubMed]
- Cazares, D.; Cazares, A.; Figueroa, W.; Guarneros, G.; Edwards, R.A.; Vinuesa, P. A Novel Group of Promiscuous Podophages Infecting Diverse Gammaproteobacteria from River Communities Exhibits Dynamic Intergenus Host Adaptation. mSystems 2021, 6, e00773-20. [Google Scholar] [CrossRef] [PubMed]
- Uhrynowski, W.; Decewicz, P.; Dziewit, L.; Radlinska, M.; Krawczyk, P.S.; Lipinski, L.; Adamska, D.; Drewniak, L. Analysis of the Genome and Mobilome of a Dissimilatory Arsenate Reducing Aeromonas sp. O23A Reveals Multiple Mechanisms for Heavy Metal Resistance and Metabolism. Front. Microbiol. 2017, 8, 936. [Google Scholar] [CrossRef]
- Bujak, K.; Decewicz, P.; Rosinska, J.M.; Radlinska, M. Genome Study of a Novel Virulent Phage vB_SspS_KASIA and Mu-like Prophages of Shewanella sp. M16 Provides Insights into the Genetic Diversity of the Shewanella Virome. Int. J. Mol. Sci. 2021, 22, 11070. [Google Scholar] [CrossRef]
- Bujak, K.; Decewicz, P.; Kaminski, J.; Radlinska, M. Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine. Int. J. Mol. Sci. 2020, 21, 6709. [Google Scholar] [CrossRef]
- Golec, P.; Karczewska-Golec, J.; Łos, M.; Węgrzyn, G. Bacteriophage T4 can produce progeny virions in extremely slowly growing Escherichia coli host: Comparison of a mathematical model with the experimental data. FEMS Microbiol. Lett. 2014, 351, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, H.; Pinto, G.; Oliveira, A.; Oliveira, C.; Faustino, M.; Briers, Y.; Domingues, L.; Azeredo, J. Characterization and genome sequencing of a Citrobacter freundii phage CfP1 harboring a lysin active against multidrug-resistant isolates. Appl. Microbiol. Biotechnol. 2016, 100, 10543–10553. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 2018, 7, 1338. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2016, 45, D535–D542. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Söding, J. Fast and accurate automatic structure prediction with HHpred. Proteins 2009, 77, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Sonnhammer, E.L.L.; Paladin, L.; Richardson, L.J.; Finn, R.D.; Bateman, A. Pfam: The protein families database in 2021. Nucleic Acids Res. 2022, 49, D412–D419. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2022, 49, D412–D419. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.J.; Todeschini, T.C.; Wu, Y.; Perry, B.J.; Ronson, C.W.; Fineran, P.C.; Nobrega, F.L.; Jackson, S.A. Identification and classification of antiviral defence systems in bacteria and archaea with PADLOC reveals new system types. Nucleic Acids Res. 2022, 49, 10868–10878. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-Scale Biases in the Current Collection of Cultured Phage Genomes. PHAGE 2021, 2, 214–223. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI Conference Weblogs Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar] [CrossRef]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a Continuous Graph Layout Algorithm for Handy Network Visualization Designed for the Gephi Software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2022, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2022, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2022, 35, 518–522. [Google Scholar] [CrossRef]
- Adamczuk, M.; Dziewit, L. Genome-based insights into the resistome and mobilome of multidrug-resistant Aeromonas sp. ARM81 isolated from wastewater. Arch. Microbiol. 2017, 199, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2022, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Uhrynowski, W.; Debiec, K.; Sklodowska, A.; Drewniak, L. The role of dissimilatory arsenate reducing bacteria in the biogeochemical cycle of arsenic based on the physiological and functional analysis of Aeromonas sp. O23A. Sci. Total Environ. 2017, 598, 680–689. [Google Scholar] [CrossRef]
- Roberts, R.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef]
- Gordeeva, J.; Morozova, N.; Sierro, N.; Isaev, A.; Sinkunas, T.; Tsvetkova, K.; Matlashov, M.; Truncaite, L.; Morgan, R.; Ivanov, N.; et al. BREX system of Escherichia coli distinguishes self from non-self by methylation of a specific DNA site. Nucleic Acids Res. 2019, 47, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Mestre, M.R.; González-Delgado, A.; Gutierrez-Rus, L.; Martínez-Abarca, F.; Toro, N. Systematic prediction of genes functionally associated with bacterial retrons and classification of the encoded tripartite systems. Nucleic Acids Res. 2020, 48, 12632–12647. [Google Scholar] [CrossRef]
- Millman, A.; Bernheim, A.; Stokar-Avihail, A.; Fedorenko, T.; Voichek, M.; Leavitt, A.; Oppenheimer-Shaanan, Y.; Sorek, R. Bacterial Retrons Function In Anti-Phage Defense. Cell 2020, 183, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, N.; Morehouse, B.; Millman, A.; Stokar-Avihail, A.; Avraham, C.; Fedorenko, T.; Yirmiya, E.; Herbst, E.; Brandis, A.; Mehlman, T.; et al. Cyclic CMP and cyclic UMP mediate bacterial immunity against phages. Cell 2021, 184, 5728–5739. [Google Scholar] [CrossRef] [PubMed]
- Cavicchioli, R. On the concept of a psychrophile. ISME J. 2016, 10, 793–795. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, R.; Xu, M.; Liu, Y.; Zhu, X.; Qiu, J.; Liu, Q.; He, P.; Li, Q. A Novel Polysaccharide Depolymerase Encoded by the Phage SH-KP152226 Confers Specific Activity against Multidrug-Resistant Klebsiella pneumoniae via Biofilm Degradation. Front. Microbiol. 2019, 10, 2768. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Yang, X.; Euler, C.W.; Han, X.; Liu, J.; Hossen, I.; Zhou, Y.L.; Li, J. Application of a novel phage ZPAH7 for controlling multidrug-resistant Aeromonas hydrophila on lettuce and reducing biofilms. Food Control 2021, 122, 107785. [Google Scholar] [CrossRef]
- Liu, J.; Gao, S.; Dong, Y.; Lu, C.; Liu, Y. Isolation and characterization of bacteriophages against virulent Aeromonas hydrophila. BMC Microbiol. 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Islam, M.S.; Raz, A.; Liu, Y.; Elbassiony, K.R.A.; Dong, X.; Zhou, P.; Zhou, Y.; Jinquan, L. Complete Genome Sequence of Aeromonas Phage ZPAH7 with Halo Zones, Isolated in China | Microbiology Resource Announcements. Microbiol. Resour. Announc. 2021, 8, e01678-18. [Google Scholar]
- Xu, Z.; Jin, P.; Zhou, X.; Zhang, Y.; Wang, Q.; Liu, X.; Shao, S.; Liu, Q.; Dozois, C.M. Isolation of a Virulent Aeromonas salmonicida subsp. masoucida Bacteriophage and Its Application in Phage Therapy in Turbot (Scophthalmus maximus). Appl. Environ. Microbiol. 2021, 87, e0146821. [Google Scholar] [CrossRef]
- Cornelissen, A.; Ceyssens, P.-J.; T’Syen, J.; Van Praet, H.; Noben, J.-P.; Shaburova, O.V.; Krylov, V.N.; Volckaert, G.; Lavigne, R. The T7-Related Pseudomonas putida Phage φ15 Displays Virion-Associated Biofilm Degradation Properties. PLoS ONE 2011, 6, e18597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlebicki, A.; Zielenkiewicz, U.; Wilczek, A.M. Fungi are not involved in biofilm formation on rock wall in subterranean arsenic mine in Poland. Nova Hedwig. 2014, 99, 255–269. [Google Scholar] [CrossRef]
- Przylibski, T.A. Radon and its daughter products behaviour in the air of an underground tourist route in the former arsenic and gold mine in Złoty Stok (Sudety Mountains, SW Poland). J. Environ. Radioact. 2001, 57, 87–103. [Google Scholar] [CrossRef]
- Dekel-Bird, N.P.; Sabehi, G.; Mosevitzky, B.; Lindell, D. Host-dependent differences in abundance, composition and host range of cyanophages from the Red Sea. Environ. Microbiol. 2014, 17, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.A.; Lindell, D. Genetic hurdles limit the arms race between Prochlorococcus and the T7-like podoviruses infecting them. ISME J. 2017, 11, 1836–1851. [Google Scholar] [CrossRef] [PubMed]
- Leon-Velarde, C.G.; Kropinski, A.M.; Chen, S.; Abbasifar, A.; Griffiths, M.W.; Odumeru, J.A. Complete genome sequence of bacteriophage vB_YenP_AP5 which infects Yersinia enterocoliticaof serotype O:3. J. Virol. 2014, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Feng, C.; Zhang, L.; Chi, T.; Qi, Y.; Jia, K.; Zhang, Y.; Wei, J.; Qian, A.; Sun, W.; et al. Characterization and genome analysis of two novel Aeromonas hydrophila phages PZL-Ah1and PZL-Ah8. Arch. Virol. 2021, 167, 669–673. [Google Scholar] [CrossRef]
- Malki, K.; Kula, A.; Bruder, K.; Sible, E.; Hatzopoulos, T.; Steidel, S.; Watkins, S.C.; Putonti, C. Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. J. Virol. 2015, 12, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Betat, H.; Rammelt, C.; Mör, L.M. tRNA nucleotidyltransferases: Ancient catalysts with an unusual mechanism of polymerization. Cell. Mol. Life Sci. 2010, 67, 1447–1463. [Google Scholar] [CrossRef]
- Evseev, P.V.; Lukianova, A.A.; Shneider, M.M.; Korzhenkov, A.A.; Bugaeva, E.N.; Kabanova, A.P.; Miroshnikov, K.K.; Kulikov, E.E.; Toshchakov, S.V.; Ignatov, A.N.; et al. Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches. Microorganisms 2020, 8, 1707. [Google Scholar] [CrossRef] [PubMed]
- Hutinet, G.; Besle, A.; Son, O.; McGovern, S.; Guerois, R.; Petit, M.; Ochsenbein, F.; Lecointe, F. Sak4 of Phage HK620 Is a RecA Remote Homolog With Single-Strand Annealing Activity Stimulated by Its Cognate SSB Protein. Front. Microbiol. 2018, 9, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbins, A.T.; George, M.; Ford, M.E.; Houtz, J.M.; Pedulla, M.L.; Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Complete Genomic Sequence of the Virulent Salmonella Bacteriophage SP6. J. Bacteriol. 2004, 186, 1933–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikoulinskaia, G.V.; Odinokova, I.V.; Zimin, A.A.; Lysanskaya, V.Y.; Feofanov, S.A.; Stepnaya, O.A. Identification and characterization of the metal ion-dependent l-alanoyl-d-glutamate peptidase encoded by bacteriophage T5. FEBS J. 2009, 276, 7329–7342. [Google Scholar] [CrossRef] [PubMed]
- Lukianova, A.A.; Shneider, M.M.; Evseev, P.V.; Shpirt, A.M.; Bugaeva, E.N.; Kabanova, A.P.; Obraztsova, E.A.; Senchenkova, S.N.; Shashkov, A.S.; Shashkov, S.V.; et al. Morphologically Different Pectobacterium brasiliense Bacteriophages PP99 and PP101: Deacetylation of O-Polysaccharide by the Tail Spike Protein of Phage PP99 Accompanies the Infection. Front. Microbiol. 2020, 10, 3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabiey, M.; Roy, S.R.; Holtappels, D.; Franceschetti, L.; Quilty, B.J.; Creeth, R.; Sundin, G.W.; Wagemans, J.; Lavigne, R.; Jackson, R.W. Phage biocontrol to combat Pseudomonas syringae pathogens causing disease in cherry. Microb. Biotechnol. 2020, 13, 1428–1445. [Google Scholar] [CrossRef]
- Popova, A.; Lavysh, D.; Klimuk, E.; Edelstein, M.; Bogun, A.; Shneider, M.; Goncharov, A.; Leonov, S.; Severinov, K. Novel Fri1-like Viruses Infecting Acinetobacter baumannii-vB_AbaP_AS11 and vB_AbaP_AS12-Characterization, Comparative Genomic Analysis, and Host-Recognition Strategy. Viruses 2017, 9, 188. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, H.; Costa, A.; Konstantinides, N.; Ferreira, A.; Akturk, E.; Sillankorva, S.; Nemec, A.; Shneider, M.; Dötsch, A.; Azeredo, J. Ability of phages to infect Acinetobacter calcoaceticus-Acinetobacter baumannii complex species through acquisition of different pectate lyase depolymerase domains. Environ. Microbiol. 2017, 19, 5060–5077. [Google Scholar] [CrossRef] [Green Version]
- Timoshina, O.Y.; Shneider, M.M.; Evseev, P.V.; Shchurova, A.S.; Shelenkov, A.A.; Mikhaylova, Y.V.; Sokolova, O.S.; Kasimova, A.A.; Arbatsky, N.P.; Dmitrenok, A.S.; et al. Novel Acinetobacter baumannii Bacteriophage Aristophanes Encoding Structural Polysaccharide Deacetylase. Viruses 2021, 13, 1688. [Google Scholar] [CrossRef]
- Silva, J.B.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Lemire, S.; Pires, D.; Lu, T. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.; Kropinski, A.; Alfernas-Zerbini, P.; Buttimer, C.; Lavigne, R.; Bister, J.R.; Tolstoy, I.; Morozova, V.V.; Babkin, I.V.; Kozlova, Y.N.; et al. Create One New Family (Autographiviridae) including Nine Subfamilies and One Hundred and Thirty-Two Genera in the Order Caudovirales. Available online: https://ictv.global/ICTV/proposals/2019.103B.zip (accessed on 17 May 2020).
- Adriaenssens, E.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Chen, F.; Chu, X.; Zhang, H.; Luo, H.; Qin, F.; Zhai, Z.; Yang, M.; Sun, J.; Zhao, Y. Diverse, Abundant, and Novel Viruses Infecting the Marine Roseobacter RCA Lineage. mSystems 2019, 4, e00494-19. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Qin, F.; Zhang, R.; Giovannoni, S.; Zhang, Z.; Sun, J.; Du, S.; Rensing, C. Pelagiphages in the Podoviridae family integrate into host genomes. Environ. Microbiol. 2019, 21, 1989–2001. [Google Scholar] [CrossRef]
- Buchholz, H.H.; Michelsen, M.L.; Bolaños, L.M.; Browne, E.; Allen, M.J.; Temperton, B. Efficient dilution-to-extinction isolation of novel virus–host model systems for fastidious heterotrophic bacteria. ISME J. 2021, 15, 1585–1598. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Chen, F.; Lennon, J.T.; Wilhelm, S.W. Complete Genome Sequence of Cyanobacterial Siphovirus KBS2A. Genome Announc. 2013, 1, e00472-13. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wang, K.; Jiao, N.; Chen, F. Genome sequences of siphoviruses infecting marine Synechococcus unveil a diverse cyanophage group and extensive phage-host genetic exchanges. Environ. Microbiol. 2012, 14, 540–558. [Google Scholar] [CrossRef]
- Zhang, D.; He, Y.; Gin, K.Y. Genomic Characterization of a Novel Freshwater Cyanophage Reveals a New Lineage of Cyanopodovirus. Front. Microbiol. 2022, 12, 768868. [Google Scholar] [CrossRef]
- Zhang, D.; He, Y.; Gin, K.Y. Novel Freshwater Cyanophages Provide New Insights into Evolutionary Relationships between Freshwater and Marine Cyanophages. Microbiol. Spectr. 2021, 9, e00593-21. [Google Scholar] [CrossRef]
- Cao, Y.; Li, S.; Wang, D.J.Z.; Xu, L.; Liu, H.; Lu, T.; Mou, Z. Genomic characterization of a novel virulent phage infecting the Aeromonas hydrophila isolated from rainbow trout (Oncorhynchus mykiss). Virus Res. 2019, 273, 197764. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Vandenheuvel, D.; Kropinski, A.; Mast, J.; De Vos, D.; Verbeken, G.; Noben, J.-P.; Lavigne, R.; Vaneechoutte, M.; Pirnay, J.-P. Characterization of Newly Isolated Lytic Bacteriophages Active against Acinetobacter baumannii. PLoS ONE 2014, 9, e104853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.N.; Lin, J.W.; Weng, S.F.; Tseng, Y.H. Genomic characterization of the intron-containing T7-like phage phiL7 of Xanthomonas campestris. Appl. Environ. Microbiol. 2009, 75, 7828–7837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreienbaum, M.; Dörrich, K.A.; Brandt, D.; Schmid, E.N.; Leonhard, T.; Hager, F.; Brenzinger, S.; Hahn, J.; Glatter, T.; Ruwe, M.; et al. Isolation and Characterization of Shewanella Phage Thanatos Infecting and Lysing Shewanella oneidensis and Promoting Nascent Biofilm Formation. Front. Microbiol. 2021, 11, 573260. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Matsuura, T.; Ohara, T.; Azegami, K. Bacteriophage OP1, lytic for Xanthomonas oryzae pv. oryzae, changes its host range by duplication and deletion of the small domain in the deduced tail fiber gene. J. Gen. Plant Pathol. 2006, 72, 111–118. [Google Scholar] [CrossRef]
- Lee, C.-N.; Hu, R.-M.; Chow, T.-Y.; Lin, J.-W.; Chen, H.-Y.; Tseng, Y.-H.; Weng, S.-F. Comparison of Genomes of Three Xanthomonas oryzae Bacteriophages. BMC Genom. 2007, 8, 442. [Google Scholar] [CrossRef] [Green Version]
- Ahern, S.J.; Das, M.; Bhowmick, T.S.; Young, R.; Gonzalez, C.F. Characterization of Novel Virulent Broad-Host-Range Phages of Xylella fastidiosa and Xanthomonas. J. Bacteriol. 2014, 196, 459–471. [Google Scholar] [CrossRef]
- Colangelo-Lillis, J.R.; Deming, J.W. Genomic analysis of cold-active Colwelliaphage 9A and psychrophilic phage–host interactions. Extremophiles 2012, 17, 99–114. [Google Scholar] [CrossRef]
- Li, P.; Zhang, Y.; Yan, F.; Zhou, X. Characteristics of a Bacteriophage, vB_Kox_ZX8, Isolated From Clinical Klebsiella oxytoca and Its Therapeutic Effect on Mice Bacteremia. Front. Microbiol. 2021, 12, 763136. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; Van Vaerenbergh, J.; Maes, M.; De Proft, M.; Lavigne, R. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses”. Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Hu, Y.; Mizan, F.R.; Yan, T.; Ishatur, N.; Zhou, Y.; Li, J. Characterization of Salmonella Phage LPST153 That Effectively Targets Most Prevalent Salmonella Serovars. Microorganisms 2020, 8, 1089. [Google Scholar] [CrossRef] [PubMed]
- Petrzik, K.; Kmoch, M.; Brázdová, S.; Ševčík, R. Complete genome sequences of novel Berlinvirus and novel Certrevirus lytic for Pectobacterium sp. causing soft rot and black leg disease of potato. Virus Genes 2021, 57, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Heu, S.; Park, J.; Roh, E. Genomic characterization of bacteriophage vB_PcaP_PP2 infecting Pectobacterium carotovorum subsp. carotovorum, a new member of a proposed genus in the subfamily Autographivirinae. Arch. Virol. 2017, 162, 2441–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, H.; Maciejewska, B.; Latka, A.; Majkowska-Skrobek, G.; Hellstrand, M.; Melefors, Ö.; Wang, J.-T.; Kropinski, A.M.; Drulis-Kawa, Z.; Nilsson, A.S. A Suggested New Bacteriophage Genus, “Kp34likevirus”, within the Autographivirinae Subfamily of Podoviridae. Viruses 2015, 7, 1804–1822. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, S.; Rousseau, G.M.; Lbrie, S.J.; Kourda, R.S.; Tremblay, D.M.; Moineau, S.; Slama, K.B. Characterization of Five Podoviridae Phages Infecting Citrobacter freundii. Front. Microbiol. 2016, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drulis-Kawa, Z.; Mackiewicz, P.; Kęsik-Szeloch, A.; Maciaszczyk-Dziubinska, E.; Weber-Dąbrowska, B.; Dorotkiewicz-Jach, A.; Augustyniak, D.; Majkowska-Skrobek, G.; Bocer, T.; Empel, J.; et al. Isolation and characterization of KP34—A novel φKMV-like bacteriophage for Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2011, 90, 1333–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Jang, H.B.; Kim, K.S.; Kim, T.H.; Im, S.P.; Kim, S.W.; Lazarte, J.M.; Kim, J.S.; Jung, T.S. Complete Genomic and Lysis-Cassette Characterization of the Novel Phage, KBNP1315, which Infects Avian Pathogenic Escherichia coli (APEC). PLoS ONE 2015, 10, e0142504. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MR7 | MR16 | MR19 | |
|---|---|---|---|
| Number of contigs | 54 | 97 | 61 |
| Sequenced genome size (bp) | 4,822,910 | 4,655,773 | 4,883,405 |
| GC content (%) | 60.4 | 60.9 | 60.2 |
| Number of predicted genes | 4364 | 4236 | 4367 |
| Number of putative virulence factors Antibiotic Resistance/Virulence Factor | 56/59 | 41/49 | 51/49 |
| Number of prophages | 1 | 3 | 1 |
| Number of genes encoding transposases | 14 | 52 | 22 |
| Bolek | Lolek | Tola | ||||
|---|---|---|---|---|---|---|
| R2A | LB | R2A | LB | R2A | LB | |
| Latency period (min) | 60 | 60 | 60 | 60 | 70 | 60 |
| Rise period (min) | 70 | 70 | 20 | 20 | 20 | 30 |
| Burst size (PFU/IC) | 73 ± 8 | 70 ± 10 | 68 ± 8 | 62 ± 16 | 107 ± 8 | 115 ± 10 |
| Bolek | Lolek | Tola | |
|---|---|---|---|
| Sequenced genome size (bp) | 42,433 | 43,136 | 45,276 |
| Physical genome size (bp) | 42,747 | 43,638 | 45,907 |
| Terminal repeats size (bp) | 314 | 502 | 631 |
| GC content (%) | 52.7 | 52.6 | 47.4 |
| Number of predicted genes | 57 | 61 | 70 |
| Number of predicted gene products | 27 | 27 | 32 |
| Number of hypothetical proteins | 30 | 34 | 38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bujak, K.; Decewicz, P.; Kitowicz, M.; Radlinska, M. Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family. Viruses 2022, 14, 1016. https://doi.org/10.3390/v14051016
Bujak K, Decewicz P, Kitowicz M, Radlinska M. Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family. Viruses. 2022; 14(5):1016. https://doi.org/10.3390/v14051016
Chicago/Turabian StyleBujak, Katarzyna, Przemyslaw Decewicz, Michal Kitowicz, and Monika Radlinska. 2022. "Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family" Viruses 14, no. 5: 1016. https://doi.org/10.3390/v14051016
APA StyleBujak, K., Decewicz, P., Kitowicz, M., & Radlinska, M. (2022). Characterization of Three Novel Virulent Aeromonas Phages Provides Insights into the Diversity of the Autographiviridae Family. Viruses, 14(5), 1016. https://doi.org/10.3390/v14051016