
Studies of Infection and Experimental Reactivation by Recombinant VZV with Mutations in Virally-Encoded Small Non-Coding RNA

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Neuronal Differentiation from hESC
2.3. Generation of VZV Mutant in VZVsncRNA
2.4. Stem-Loop Taqman qPCR for VZVsncRNA Quantification and Detection
2.5. qRT-PCR for Viral Transcripts
2.6. Latency/Reactivation Experiments
2.7. Multistep Growth-Kinetics
3. Results
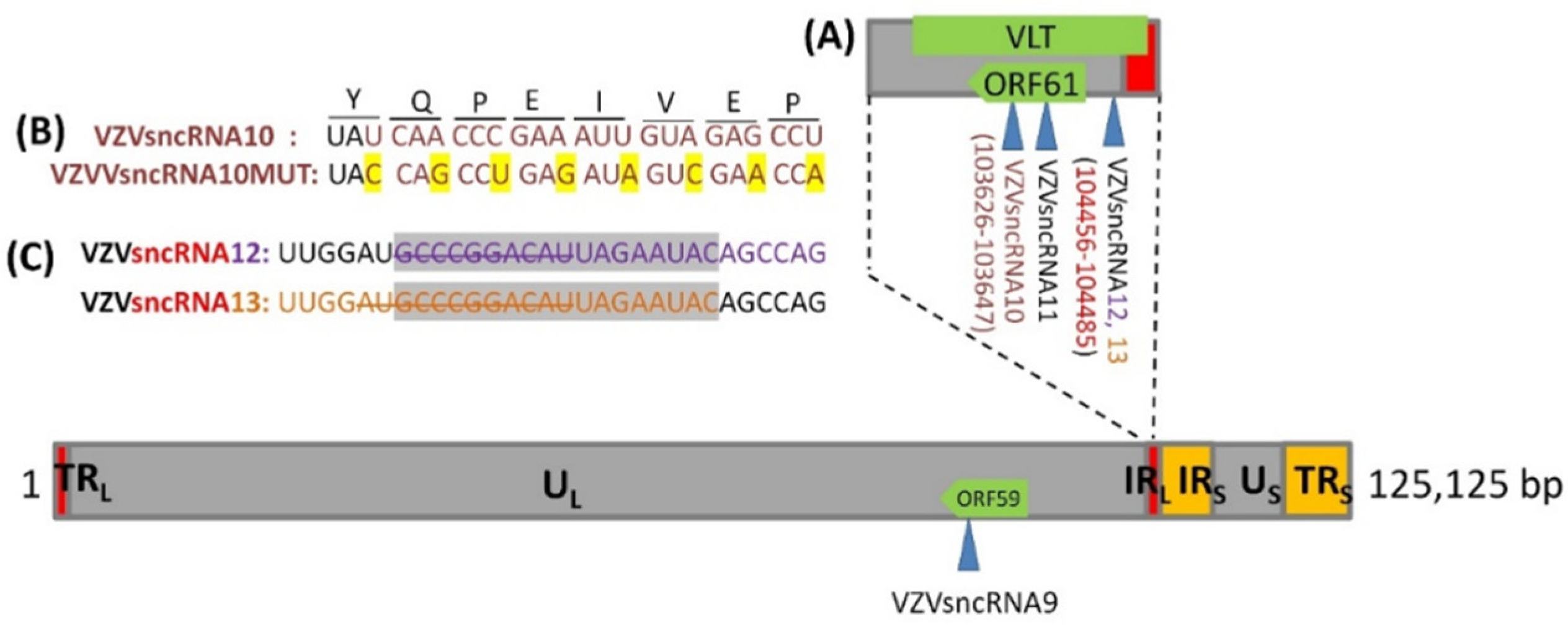
3.1. Construction of VZV with Synonymous Mutation of VZVsncRNA10 and Deletion in the Overlap Region between VZVsncRNA 12&13
3.2. Growth Rates of VZVsnc10MUT and VZVsnc12-13DEL Are Similar to Those of Their Parental Virus in ARPE-19 Cells
3.3. VZVsnc10MUT and VZVsnc12-13DEL Can Productively Infect Human Embryonic Stem Cell-Derived Neurons
3.4. VZV Mutated for VZVsncRNA Derived from the mRNA for ORF61 Can Establish Experimental Latency in hESC-Derived Neurons
3.5. VZV Mutated in VZVsncRNA10 or Loss of VZVsncRNA12/13 Display Impaired Reactivation from Latency
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Arvin, A.M. The pathogenesis of varicella-zoster virus neurotropism and infection. In Neurotropic Viral Infections; Cambridge University Press: Cambridge, UK, 2008; pp. 225–250. [Google Scholar]
- Takahashi, M. Development of a live varicella vaccine--past and future. Jpn. J. Infect. Dis. 2000, 53, 47–55. [Google Scholar] [PubMed]
- Watson, S.; Mercier, S.; Bye, C.; Wilkinson, J.; Cunningham, A.L.; Harman, A.N. Determination of suitable housekeeping genes for normalisation of quantitative real time PCR analysis of cells infected with human immunodeficiency virus and herpes viruses. Virol. J. 2007, 4, 130. [Google Scholar] [CrossRef] [PubMed]
- Sloutskin, A.; Goldstein, R.S. Laboratory preparation of Varicella-Zoster Virus: Concentration of virus-containing supernatant, use of a debris fraction and magnetofection for consistent cell-free VZV infections. J. Virol. Methods 2014, 206, 128–132. [Google Scholar] [CrossRef]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An in vitro model of latency and reactivation of varicella zoster virus in human stem cell-derived neurons. PLoS Pathog. 2015, 11, e1004885. [Google Scholar] [CrossRef]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Depledge, D.P.; Rajbhandari, L.; Lenac Rovis, T.; Jonjic, S.; Breuer, J.; Venkatesan, A.; Verjans, G.M.G.M.; Sadaoka, T. Varicella-zoster virus VLT-ORF63 fusion transcript induces broad viral gene expression during reactivation from neuronal latency. Nat. Commun. 2020, 11, 6324. [Google Scholar] [CrossRef]
- Han, Z.; Liu, X.; Chen, X.; Zhou, X.; Du, T.; Roizman, B.; Zhou, G. miR-H28 and miR-H29 expressed late in productive infection are exported and restrict HSV-1 replication and spread in recipient cells. Proc. Natl. Acad. Sci. USA 2016, 113, E894–E901. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Morris, K.V. Transcriptional gene silencing in humans. Nucleic Acids Res. 2016, 44, 6505–6517. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Golani, L.; Ojha, N.K.; Borodianskiy-Shteinberg, T.; Kinchington, P.R.; Goldstein, R.S. Varicella-Zoster Virus Expresses Multiple Small Noncoding RNAs. J. Virol. 2017, 91, e01710-17. [Google Scholar] [CrossRef] [PubMed]
- Golani-Zaidie, L.; Borodianskiy-Shteinberg, T.; Bisht, P.; Das, B.; Kinchington, P.R.; Goldstein, R.S. Bioinformatically-predicted varicella zoster virus small non-coding RNAs are expressed in lytically-infected epithelial cells and neurons. Virus Res. 2019, 274, 197773. [Google Scholar] [CrossRef] [PubMed]
- Bisht, P.; Das, B.; Kinchington, P.R.; Goldstein, R.S. Varicella-Zoster Virus (VZV) Small Noncoding RNAs Antisense to the VZV Latency-Encoded Transcript VLT Enhance Viral Replication. J. Virol. 2020, 94, e00123-20. [Google Scholar] [CrossRef] [PubMed]
- Perusina Lanfranca, M.; Mostafa, H.H.; Davido, D.J. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438–454. [Google Scholar] [CrossRef]
- Das, B.; Bisht, P.; Kinchington, P.R.; Goldstein, R.S. Locked-nucleotide antagonists to varicella zoster virus small non-coding RNA block viral growth and have potential as an anti-viral therapy. Antivir. Res. 2021, 193, 105144. [Google Scholar] [CrossRef] [PubMed]
- Birenboim, R.; Markus, A.; Goldstein, R.S. Simple generation of neurons from human embryonic stem cells using agarose multiwell dishes. J. Neurosci. Methods 2013, 214, 9–14. [Google Scholar] [CrossRef]
- Martin, D.P.; Wallace, T.L.; Johnson, E.M. Cytosine arabinoside kills postmitotic neurons in a fashion resembling trophic factor deprivation: Evidence that a deoxycytidine-dependent process may be required for nerve growth factor signal transduction. J. Neurosci. 1990, 10, 184–193. [Google Scholar] [CrossRef]
- Morris, E.J.; Geller, H.M. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: Evidence for cell cycle-independent toxicity. J. Cell Biol. 1996, 134, 757–770. [Google Scholar] [CrossRef]
- Tischer, B.K.; Kaufer, B.B.; Sommer, M.; Wussow, F.; Arvin, A.M.; Osterrieder, N. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J. Virol. 2007, 81, 13200–13208. [Google Scholar] [CrossRef]
- Erazo, A.; Yee, M.B.; Osterrieder, N.; Kinchington, P.R. Varicella-zoster virus open reading frame 66 protein kinase is required for efficient viral growth in primary human corneal stromal fibroblast cells. J. Virol. 2008, 82, 7653–7665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mohammadi-Yeganeh, S.; Paryan, M.; Mirab Samiee, S.; Soleimani, M.; Arefian, E.; Azadmanesh, K.; Mostafavi, E.; Mahdian, R.; Karimipoor, M. Development of a robust, low cost stem-loop real-time quantification PCR technique for miRNA expression analysis. Mol. Biol. Rep. 2013, 40, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Grigoryan, S.; Sloutskin, A.; Yee, M.B.; Zhu, H.; Yang, I.H.; Thakor, N.V.; Sarid, R.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: Direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J. Virol. 2011, 85, 6220–6233. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques 2006, 40, 191–197. [Google Scholar]
- Wu, B.W.; Yee, M.B.; Goldstein, R.S.; Kinchington, P.R. Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71. Viruses 2022, 14, 378. [Google Scholar] [CrossRef]
- Sadaoka, T.; Depledge, D.P.; Rajbhandari, L.; Venkatesan, A.; Breuer, J.; Cohen, J.I. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc. Natl. Acad. Sci. USA 2016, 113, E2403–E24012. [Google Scholar] [CrossRef]
- Bhela, S.; Rouse, B.T. Are miRNAs critical determinants in herpes simplex virus pathogenesis? Microbes Infect. 2018, 20, 461–465. [Google Scholar] [CrossRef]
- Barrozo, E.R.; Nakayama, S.; Singh, P.; Vanni, E.A.H.; Arvin, A.M.; Neumann, D.M.; Bloom, D.C. Deletion of Herpes Simplex Virus 1 MicroRNAs miR-H1 and miR-H6 Impairs Reactivation. J. Virol. 2020, 94, e00639-20. [Google Scholar] [CrossRef]
- Tormanen, K.; Wang, S.; Matundan, H.H.; Yu, J.; Jaggi, U.; Ghiasi, H. Herpes Simplex Virus 1 Small Noncoding RNAs 1 and 2 Activate the Herpesvirus Entry Mediator Promoter. J. Virol. 2022, 96, e0198521. [Google Scholar] [CrossRef]
- Wang, W.; Pan, D.; Fu, W.; Ye, X.; Han, J.; Yang, L.; Jia, J.; Liu, J.; Zhu, R.; Zhang, Y.; et al. Development of a skin- and neuro-attenuated live vaccine for varicella. Nat. Commun. 2022, 13, 824. [Google Scholar] [CrossRef]
- Selariu, A.; Cheng, T.; Tang, Q.; Silver, B.; Yang, L.; Liu, C.; Ye, X.; Markus, A.; Goldstein, R.S.; Cruz Cosme, R.S.; et al. ORF7 of Varicella Zoster Virus is a Neurotropic Factor. J. Virol. 2012, 86, 8614–8624. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.N. | Primer Name | Primer Sequence (5′→3′) |
|---|---|---|
| 1 | VZVsncRNA10MUTFW | CTTTACTCGGATGGCTTGATGATCAACTTGCGGACTGTACCAGCCTGAGATAGTCGAACCAACAAAAATGTTGATATAGGATGACGACGATAAGTAGC |
| 2 | VZVsncRNA10MUTREV | GCCCGTAAATACCTATATAGTTTAATATCAAACATTTTTGTTGGTTCGACTATCTCAGGCTGGTACAGTTCCGCAAGTCAGGGTAATGCCAGTGTTAC |
| 3 | SNC1213DelFW | GCTACCGCCCGCTAATATGGTATCCATGGTAACAACTGGCTGTATTCTACCAAACACGTAGCAGAACTGAGGATGACGACGATAAGTAGG |
| 4 | SNC1213delRev | TCACAATTTAGAACGCATGGCAGTTCTGCTACGTGTTTGGTAGAATACAGCCAGTTGTTACAGGGTAATGCCAGTGTTAC |
| ORF | FW Primer Sequence (5′→3′) | Rev Primer Sequence (5′→3′) |
|---|---|---|
| ORF31 | CCGTGGGATTATTGGTTTTG | CGACGGTTCAGTGTTTTGTG |
| ORF61 | AAAGCCTGACTTTTTGGGGT | CAAACCTGGACCTGGAAAGA |
| ORF63 | ATTGAGGCGCCGAATGTTC | CTTCACCACCATCATCAGATACG |
| GAPDH | CACATGGCCTCCAAGGAGTAA | TGAGGGTCTCTCTCTTCCTCTTG |
| VLT exon 3–4 | TGGACGATCACGGTAGTCCT | CGGAAAAACCATGCCGTGTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisht, P.; Das, B.; Borodianskiy-Shteinberg, T.; Kinchington, P.R.; Goldstein, R.S. Studies of Infection and Experimental Reactivation by Recombinant VZV with Mutations in Virally-Encoded Small Non-Coding RNA. Viruses 2022, 14, 1015. https://doi.org/10.3390/v14051015
Bisht P, Das B, Borodianskiy-Shteinberg T, Kinchington PR, Goldstein RS. Studies of Infection and Experimental Reactivation by Recombinant VZV with Mutations in Virally-Encoded Small Non-Coding RNA. Viruses. 2022; 14(5):1015. https://doi.org/10.3390/v14051015
Chicago/Turabian StyleBisht, Punam, Biswajit Das, Tatiana Borodianskiy-Shteinberg, Paul R. Kinchington, and Ronald S. Goldstein. 2022. "Studies of Infection and Experimental Reactivation by Recombinant VZV with Mutations in Virally-Encoded Small Non-Coding RNA" Viruses 14, no. 5: 1015. https://doi.org/10.3390/v14051015
APA StyleBisht, P., Das, B., Borodianskiy-Shteinberg, T., Kinchington, P. R., & Goldstein, R. S. (2022). Studies of Infection and Experimental Reactivation by Recombinant VZV with Mutations in Virally-Encoded Small Non-Coding RNA. Viruses, 14(5), 1015. https://doi.org/10.3390/v14051015