
Exercising the Sanger Sequencing Strategy for Variants Screening and Full-Length Genome of SARS-CoV-2 Virus during Alpha, Delta, and Omicron Outbreaks in Hiroshima

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Subjects
2.2. Quantification of SARS-CoV-2 Virus by qRT-PCR
2.3. Validation of the Modified In-House Primer Set hCoV-Spike-D for Amplification
2.4. Exercising Sanger Sequencing Strategy for SARS-CoV-2 Variants Screening
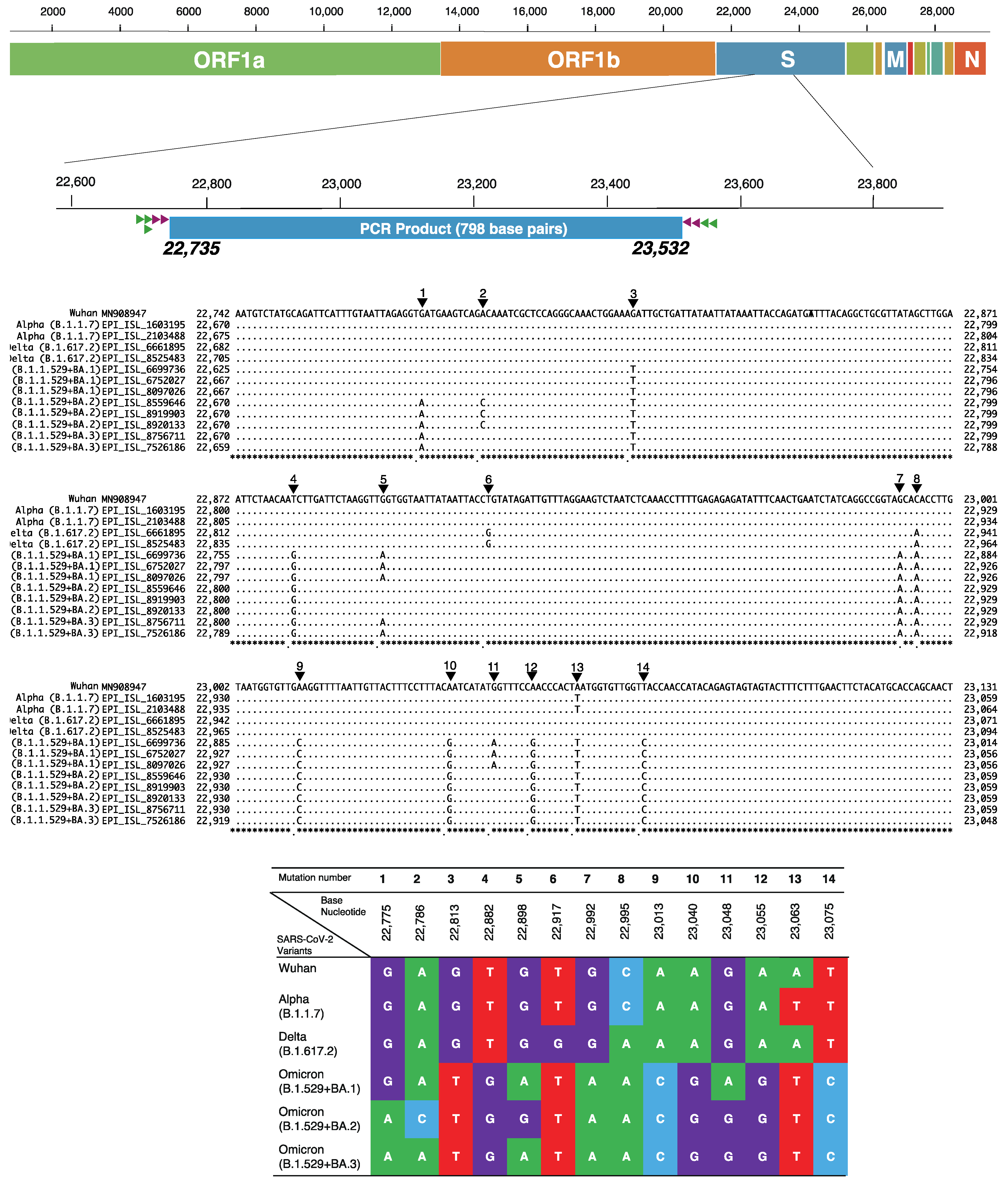
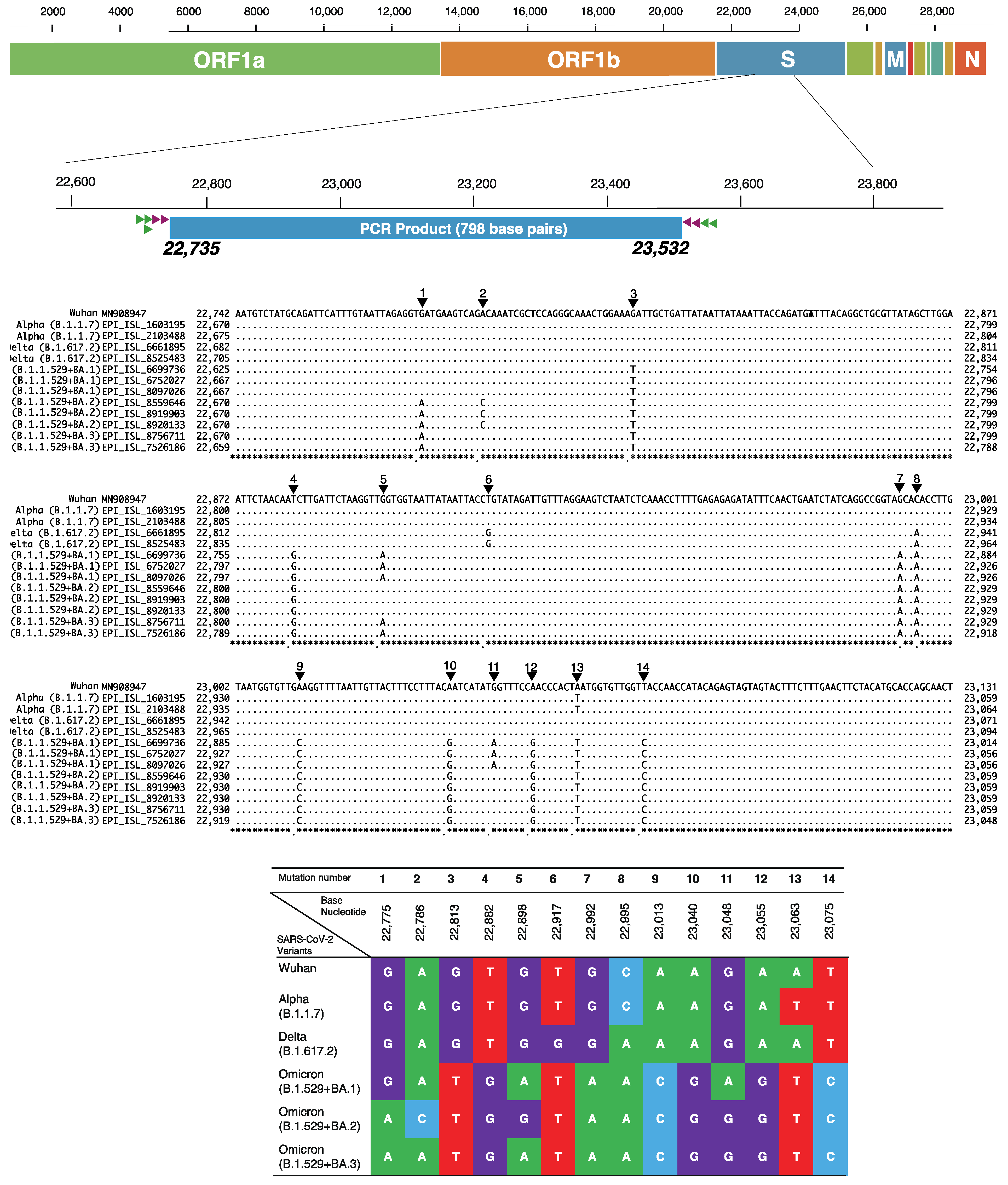
2.5. Identification of SARS-CoV-2 Variants by Unique Checkpoints of Partial Spike Gene
2.6. Full Genome Sequencing of SARS-CoV-2 Using Sanger Sequencing Method
2.7. Phylogenetic Tree of SARS-CoV-2 Full-Genome Sequences in Hiroshima
3. Results
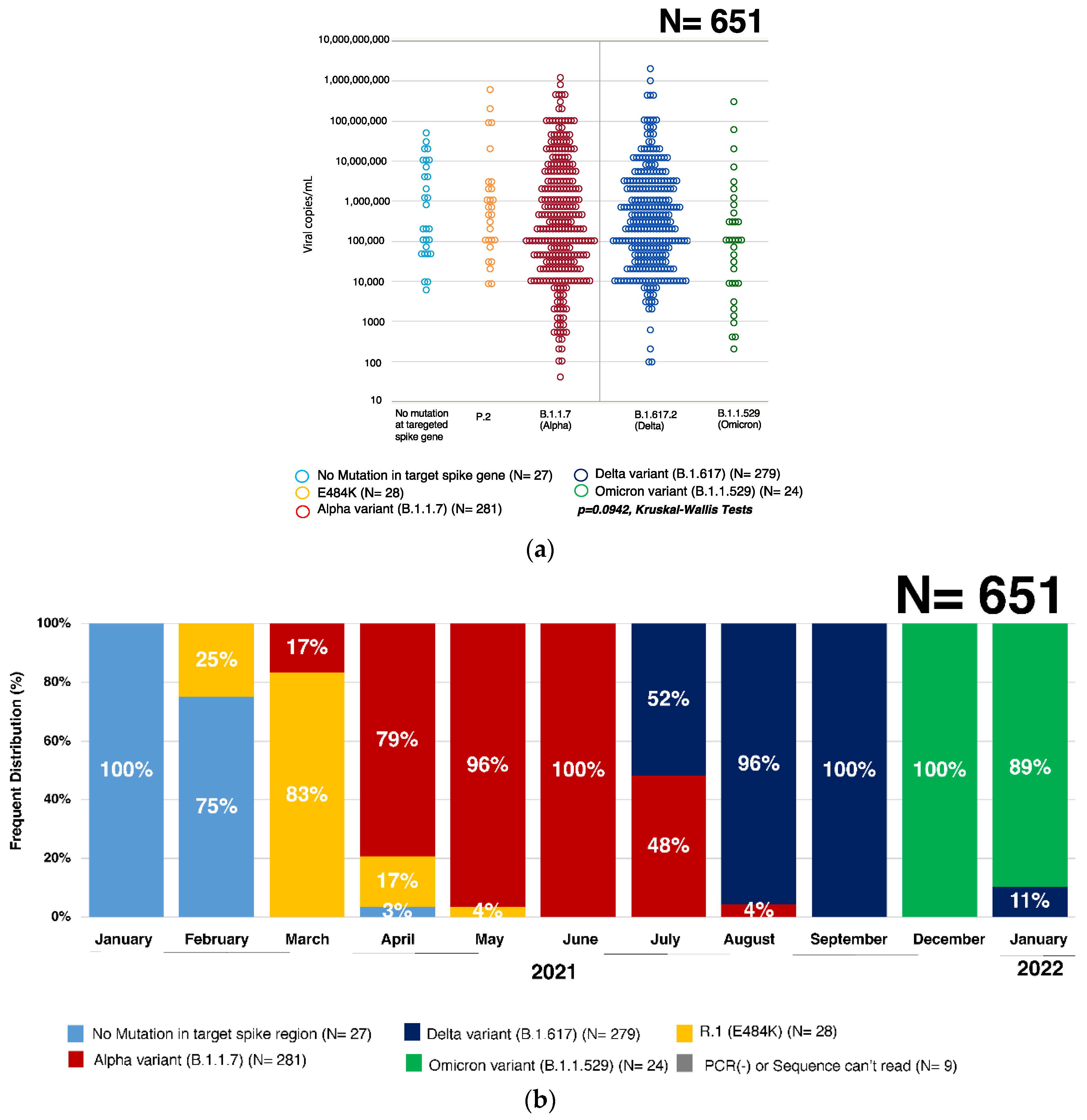
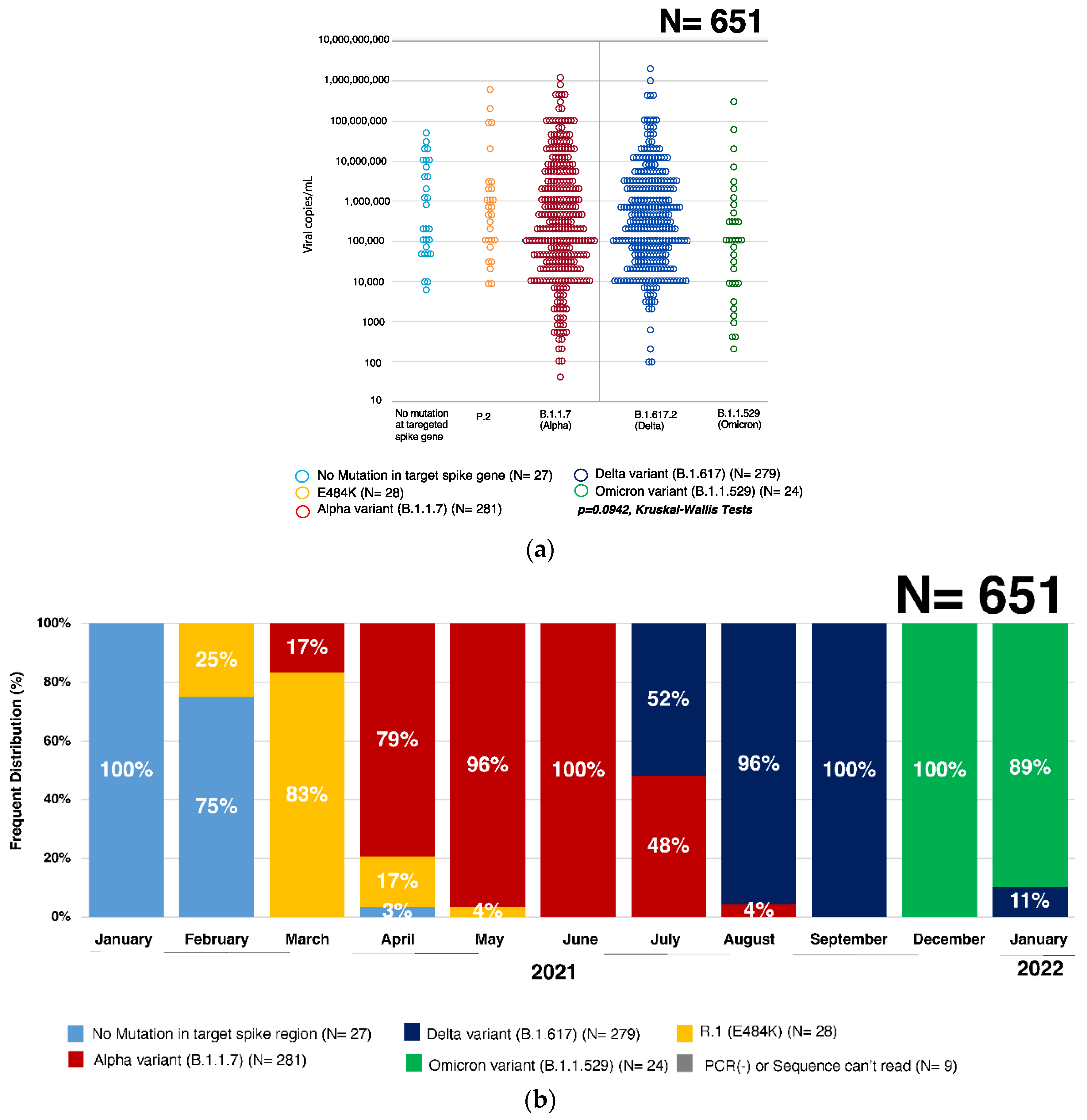
3.1. Quantification of SARS-CoV-2 Virus by qRT-PCR
3.2. Validation of the Modified In-House Primer Set hCoV-Spike-D for Amplification
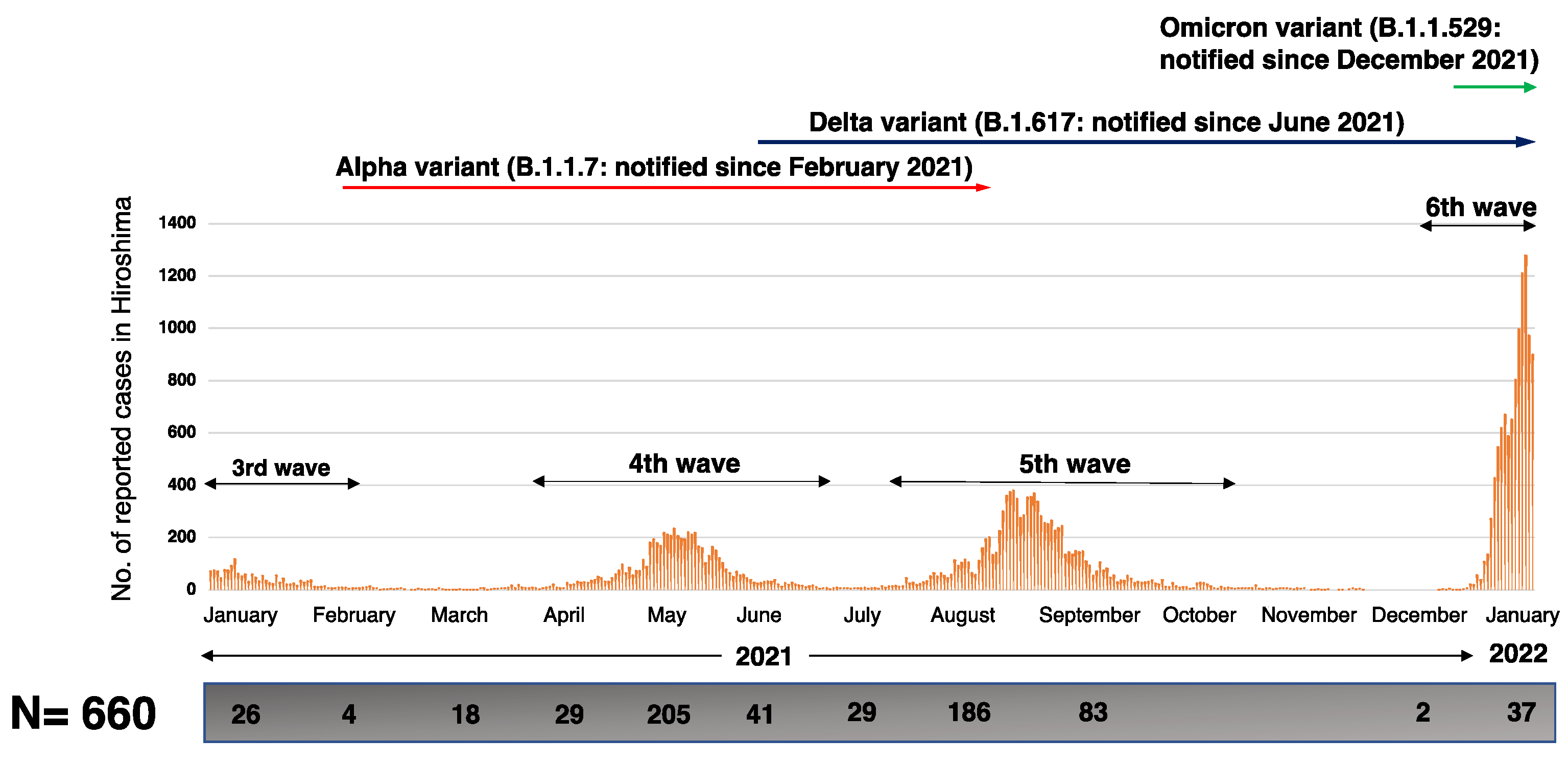
3.3. Percent Distribution of SARS-CoV-2 Variants in Hiroshima
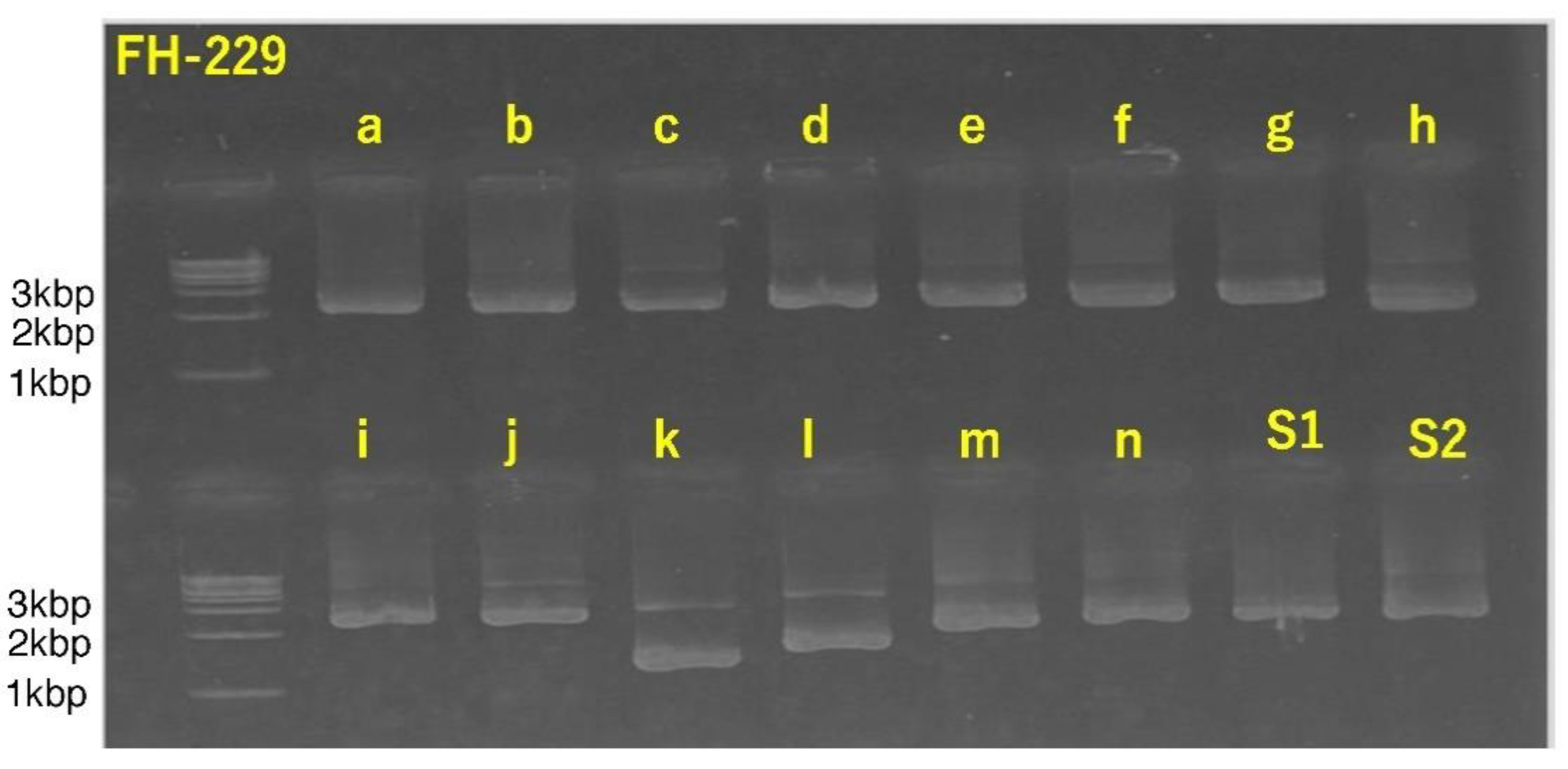
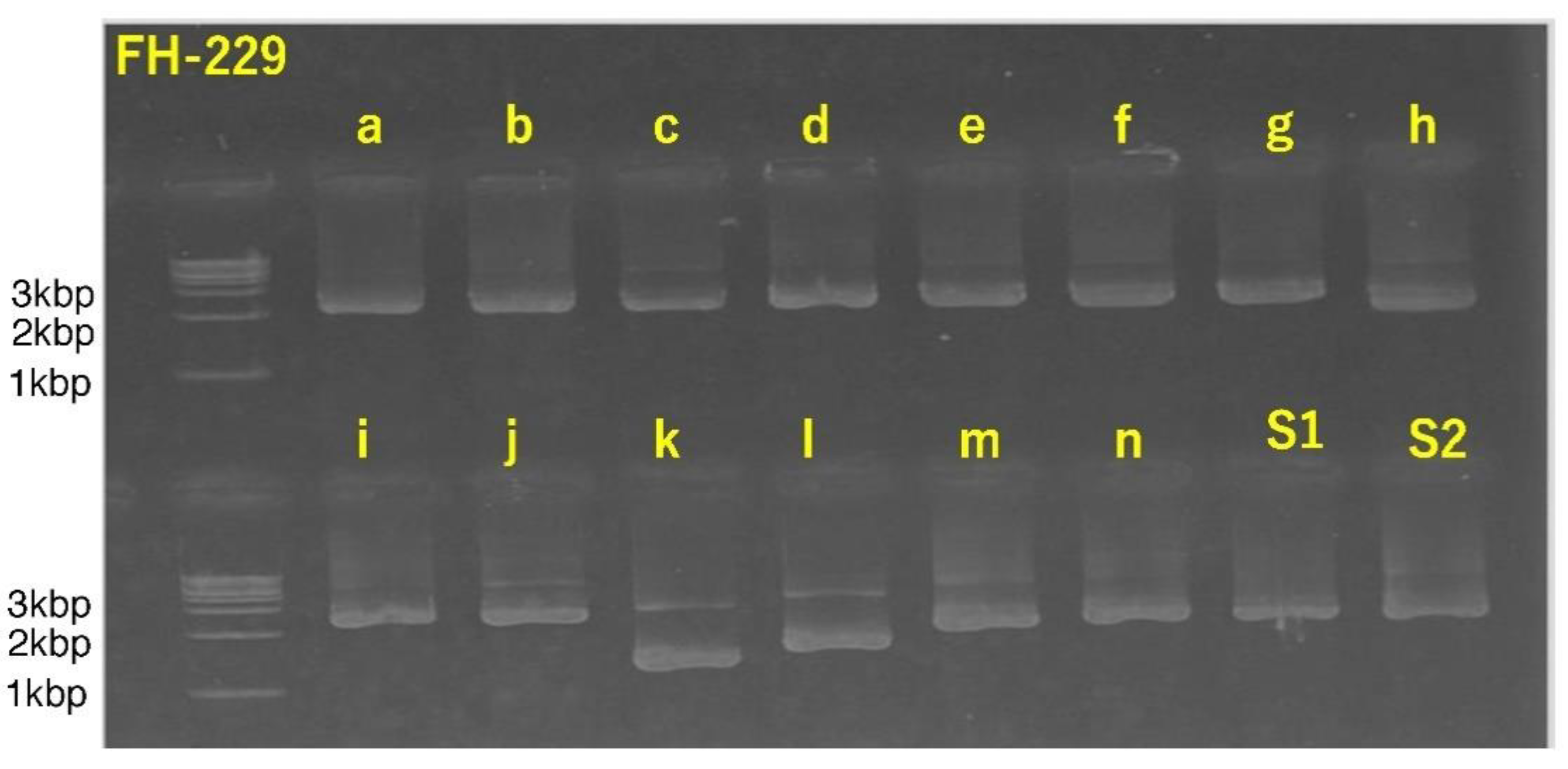
3.4. Successful Amplification of SARS-CoV-2 Full-Length Genome Using Combination of Sanger Sequencing Method and In-House-Developed Primer Library
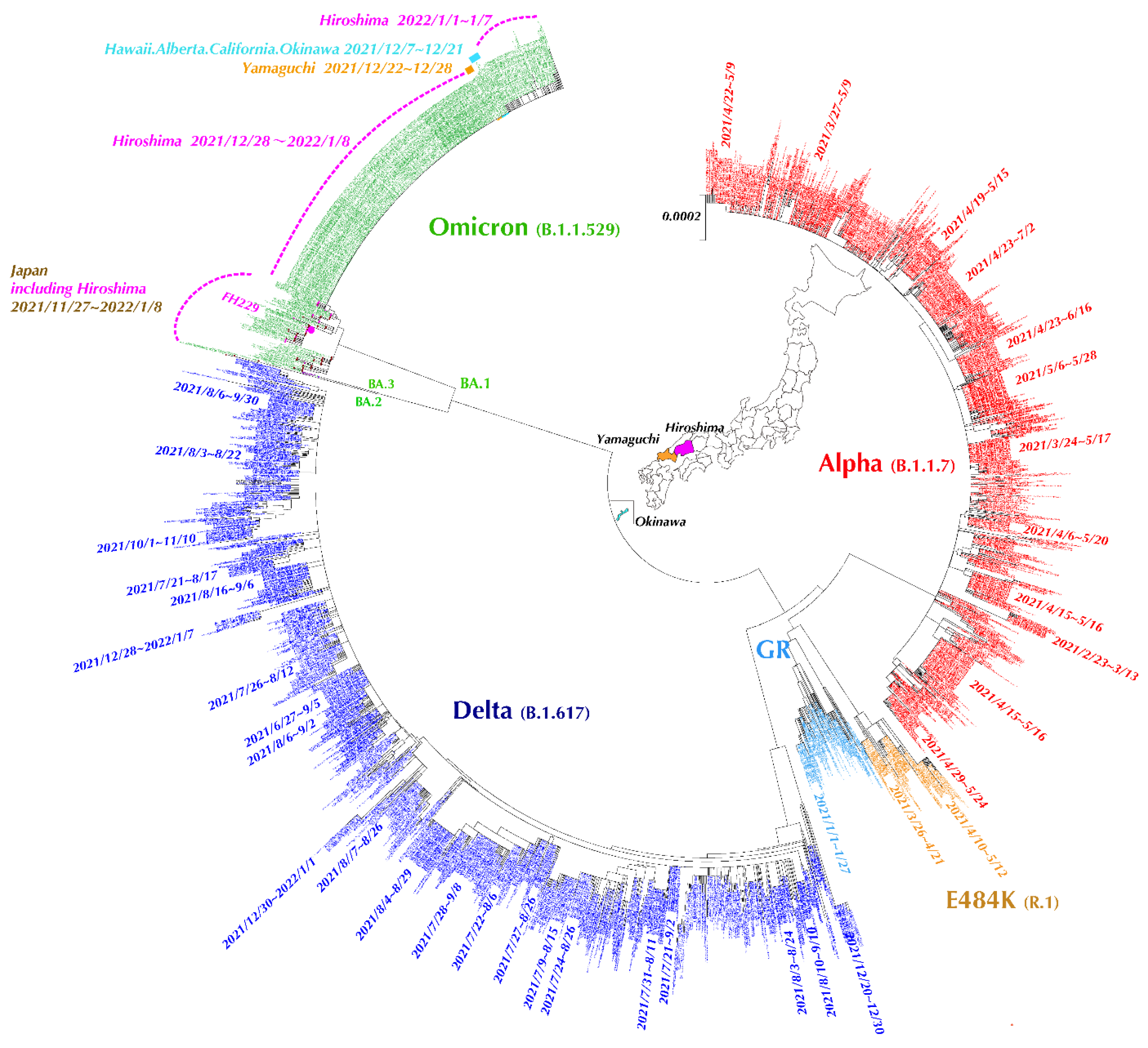
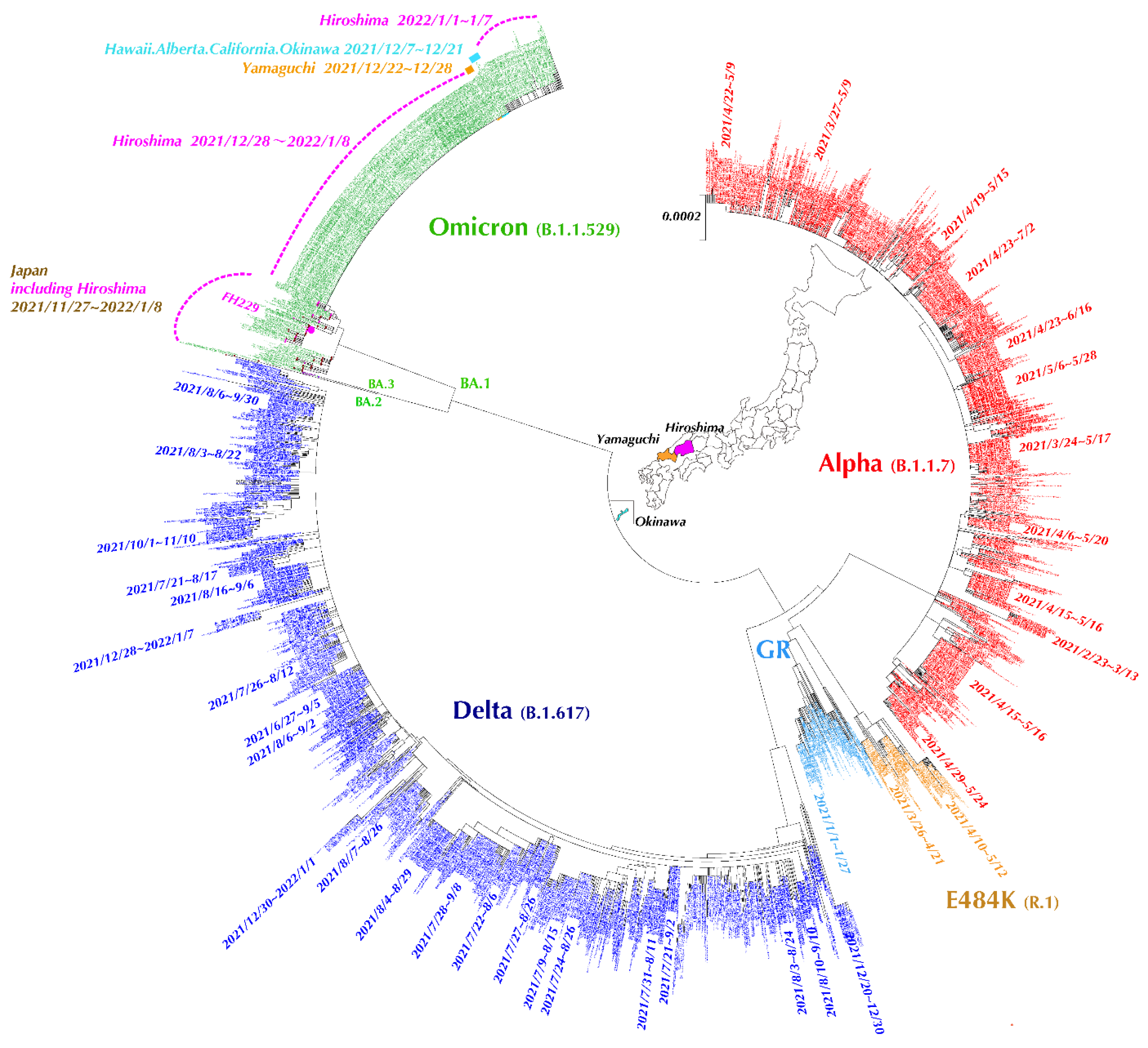
3.5. Evolutionary Analysis of SARS-CoV-2 Full-Length Genome Sequences in Hiroshima Retrieved from GISAID
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World_Health_Organization (WHO). Tracking SARS-CoV-2 Variants. 2022. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 12 March 2022).
- Bushman, M.; Kahn, R.; Taylor, B.P.; Lipsitch, M.; Hanage, W.P. Population impact of SARS-CoV-2 variants with enhanced transmissibility and/or partial immune escape. Cell 2021, 184, 6229–6242.e18. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Takahashi, K.; Nagashima, S.; E., B.; Ouoba, S.; Hussain, M.R.A.; Akita, T.; Sugiyama, A.; Sakaguchi, T.; Tahara, H.; et al. Mass Screening of SARS-CoV-2 Variants using Sanger Sequencing Strategy in Hiroshima, Japan. Sci. Rep. 2022, 12, 2419. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Nagashima, S.; E., B.; Ouoba, S.; Akita, T.; Sugiyama, A.; Ohisa, M.; Sakaguchi, T.; Tahara, H.; Ohge, H.; et al. Molecular characterization and the mutation pattern of SARS-CoV-2 during first and second wave outbreaks in Hiroshima, Japan. PLoS ONE 2021, 16, e0246383. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Kumar, S.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Hiroshima_Prefectural_Government (Office_WebPage). COVID-19 Infection Trend in Hiroshima Prefecture. Available online: https://www.city.hiroshima.lg.jp/site/english/213089.html (accessed on 12 March 2022).
- World_Health_Organization (WHO). Coronavirus Disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update. 2022. Available online: https://apps.who.int/iris/bitstream/handle/10665/352608/CoV-weekly-sitrep22Mar22-eng.pdf?sequence=1 (accessed on 12 March 2022).
- Dao Thi, V.L.; Herbst, K.; Boerner, K.; Meurer, M.; Kremer, L.P.; Kirrmaier, D.; Freistaedter, A.; Papagiannidis, D.; Galmozzi, C.; Stanifer, M.L.; et al. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 2020, 12, eabc7075. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Querci, M.; Corbisier, P.; Marchini, A.; Buttinger, G.; van den Eede, G. Silico Design of Specific Primer Sets for the Detection of B.1.1.529 SARS-CoV-2 Variant of Concern (Omicron) (Version 01); ECDC: Stockholm, Sweden, 2021. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). Methods for the Detection and Characterisation of SARS-CoV-2 Variants—First Update; ECDC; WHO Regional Office for Europe: Stockholm, Sweden, 2021. [Google Scholar]
- Park, S.; Kim, H.; Woo, K.; Kim, J.M.; Jo, H.J.; Jeong, Y.; Lee, K.H. SARS-CoV-2 Variant Screening Using a Virus-Receptor-Based Electrical Biosensor. Nano Lett. 2022, 22, 50–57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage Polarity | Primer Name | Nucleotide Position | Nucleotide Sequence (5′-3′) | |
|---|---|---|---|---|
| hCoV-Spike-D | PCR 1st Sense | 22,632S | 22,632–22,652 | GAATCAGCAACTGTGTTGCTG |
| PCR 1st Sense | 22,659S | 22,659–22,680 | CTGTCCTATATAATTCCGCATC | |
| PCR 1st Sense | 22,659S-Omi | 22,659–22,680 | CTGTCCTATATAATCTCGCACC | |
| PCR 1st Antisense | SP35AS | 23,612–23,631 | TGACTAGCTACACTACGTGC | |
| PCR 1st Antisense | SP36AS | 23,577–23,598 | TTAGTCTGAGTCTGATAACTAG | |
| PCR 2nd Sense | 22,687S | 22,687–22,708 | CACTTTTAAGTGTTATGGAGTG | |
| PCR 2nd Sense | 22,712S | 22,712–22,734 | CCTACTAAATTAAATGATCTCTG | |
| PCR 2nd Antisense | SP37AS | 23,556–23,575 | GCATATACCTGCACCAATGG | |
| PCR 2nd Antisense | SP38AS | 23,533–23,554 | TATGTCACACTCATATGAGTTG | |
| hCoV-full-a | PCR 1st Sense | 45S | 37–64 | CAACTTTCGATCTCTTGTAGATCTGTTC |
| PCR 1st Antisense | a-1-2 | 1926–1953 | TGTAAAACACGCACAGAATTTTGAGCAG | |
| PCR 2nd Sense | 85S | 77–102 | TTAAAATCTGTGTGGCTGTCACTCGG | |
| PCR 2nd Antisense | a-2-2 | 1890–1914 | CGGGAGAAAATTGATCGTACAACAC | |
| hCoV-full-b | PCR 1st Sense | b-1-1 | 1721–1743 | GCTTTTGTGGAAACTGTGAAAGG |
| PCR 1st Antisense | b-1-2 | 3878–3902 | GCTTAACTTCCTCTTTAGGAATCTC | |
| PCR 2nd Sense | b-2-1 | 1763–1785 | AAACAAATTGTTGAATCCTGTGG | |
| PCR 2nd Antisense | b-2-2 | 3838–3863 | CAACTTGCTTTTCACTCTTCATTTCC | |
| hCoV-full-c | PCR 1st Sense | c-1-1 | 3642–3666 | AAGACATTCAACTTCTTAAGAGTGC |
| PCR 1st Antisense | c-1-2 | 5830–5854 | AATAGGACCTTTGTATTCTGAGGAC | |
| PCR 2nd Sense | c-2-1 | 3683–3706 | CAGCACGAAGTTCTACTTGCACCA | |
| PCR 2nd Antisense | c-2-2 | 5750–5775 | TTATAGTGACCACACTGGTAATTACC | |
| hCoV-full-d | PCR 1st Sense | d-1-1 | 5572–5597 | GTACATGGGCACACTTTCTTATGAAC |
| PCR 1st Antisense | d-1-2 | 7764–7790 | CAGCTTTATCAAAGTAAAGATGGATGG | |
| PCR 2nd Sense | d-2-1 | 5610–5634 | GTGTTCAGATACCTTGTACGTGTGG | |
| PCR 2nd Antisense | d-2-2 | 7725–7751 | CTGTAACACTATCAACGATGTAAGAAG | |
| hCoV-full-e | PCR 1st Sense | e-1-1 | 7533–7560 | GTACAACTATTGTTAATGGTGTTAGAAG |
| PCR 1st Antisense | e-1-2 | 9729–9753 | CTCTTTAGGTAATTACTAAAGAACC | |
| PCR 2nd Sense | e-2-1 | 7570–7594 | TGTCTATGCTAATGGAGGTAAAGGC | |
| PCR 2nd Antisense | e-2-2 | 9692–9719 | GCTTTGTGGAAATACAAATGATATAAGC | |
| hCoV-full-f | PCR 1st Sense | f-1-1 | 9518–9543 | TTCCTTATGTCATTCACTGTACTCTG |
| PCR 1st Antisense | f-1-2 | 11,669–11,694 | ACACCAAGAGTCAGTCTAAAGTAGCG | |
| PCR 2nd Sense | f-2-1 | 9558–9584 | ACTCATTCTTACCTGGTGTTTATTCTG | |
| PCR 2nd Antisense | f-2-2 | 11,634–11,659 | ACAAAAGAGGCCAAAGTAACAAGTAC | |
| hCoV-full-g | PCR 1st Sense | g-1-1 | 11,459–11,483 | TCCATGTGGGCTCTTATAATCTCTG |
| PCR 1st Antisense | g-1-2 | 13,598–13,624 | CTTCGTCCTTTTCTTGGAAGCGACAAC | |
| PCR 2nd Sense | g-2-1 | 11,494–11,518 | CTACTCAGGTGTAGTTACAACTGTC | |
| PCR 2nd Antisense | g-2-2 | 13,554–13,579 | TAGCAAAACCAGCTACTTTATCATTG | |
| hCoV-full-h | PCR 1st Sense | h-1-1 | 13,384–13,407 | CGGTATGTGGAAAGGTTATGGCTG |
| PCR 1st Antisense | h-1-2 | 15,569–15,595 | ACTTATCGGCAATTTTGTTACCATCAG | |
| PCR 2nd Sense | h-2-1 | 13,418–13,441 | CAACTCCGCGAACCCATGCTTCAG | |
| PCR 2nd Antisense | h-2-2 | 15,538–15,563 | AAAAGTGCATTAACATTGGCCGTGAC | |
| hCoV-full-i | PCR 1st Sense | i-1-1 | 15,334–15,359 | ATTATGGCCTCACTTGTTCTTGCTCG |
| PCR 1st Antisense | i-1-2 | 17,506–17,533 | CTATAGTTTTCATAAGTCTACACACTCC | |
| PCR 2nd Sense | i-2-1 | 15,380–15,406 | GCTTGTCACACCGTTTCTATAGATTAG | |
| PCR 2nd Antisense | i-2-2 | 17,453–17,478 | CTTAGTTAGCAATGTGCGTGGTGCAG | |
| hCoV-full-j | PCR 1st Sense | j-1-1 | 17,278–17,304 | GTGAATTCAACATTAGAACAGTATGTC |
| PCR 1st Antisense | j-1-2 | 19,477–19,502 | GCATGATGTCTACAGACAGCACCACC | |
| PCR 2nd Sense | j-2-1 | 17,317–17,341 | AATGCATTGCCTGAGACGACAGCAG | |
| PCR 2nd Antisense | j-2-2 | 19,448–19,473 | ATTGCAACGTGTTATACACGTAGCAG | |
| hCoV-full-k | PCR 1st Sense | k-1-1 | 19,239–19,264 | ATTTGACACTAGAGTGCTATCTAACC |
| PCR 1st Antisense | k-1-2 | 21,423–21,449 | TCTTTTAAAGACATAACAGCAGTACCC | |
| PCR 2nd Sense | k-2-1 | 19,275–19,300 | TGGTTGTGATGGTGGCAGTTTGTATG | |
| PCR 2nd Antisense | k-2-2 | 21,386–21,412 | GGGGAAATTTACTCATGTCAAATAAAG | |
| hCoV-full-l | PCR 1st Sense | l-1-1 | 25,369–25,396 | ATTACATTACACATAAACGAACTTATGG |
| PCR 1st Antisense | l-1-2 | 27,416–27,442 | GCTCACAAGTAGCGAGTGTTATCAGTG | |
| PCR 2nd Sense | l-2-1 | 25,398–25,424 | TTTGTTTATGAGAATCTTCACAATTGG | |
| PCR 2nd Antisense | l-2-2 | 27,358–27,384 | ATCAATCTCCATTGGTTGCTCTTCATC | |
| hCoV-full-m | PCR 1st Sense | m-1-1 | 27,175–27,201 | CTTTGCTTGTACAGTAAGTGACAACAG |
| PCR 1st Antisense | m-1-2 | 28,771–28,797 | CTTCTGCGTAGAAGCCTTTTGGCAATG | |
| PCR 2nd Sense | m-2-1 | 27,211–27,236 | CTCGTTGACTTTCAGGTTACTATAGC | |
| PCR 2nd Antisense | m-2-2 | 28,737–28,763 | TTGAGGAAGTTGTAGCACGATTGCAGC | |
| hCoV-full-n | PCR 1st Sense | n-1-1 | 28,539–28,563 | AGAGCTACCAGACGAATTCGTGGTG |
| PCR 1st Antisense | n-1-2 | 29,854–29,882 | TTTTTTTTTTTGTCATTCTCCTAAGAAGC | |
| PCR 2nd Sense | n-2-1 | 28,595–28,621 | ATGGTATTTCTACTACCTAGGAACTGG | |
| PCR 2nd Antisense | n-2-2 | 29,826–29,852 | ATTAAAATCACATGGGGATAGCACTAC | |
| hCoV-full-S1 | PCR 1st Sense | 20,963S | 20,955–20,980 | TCTTAATGACTTTGTCTCTGATGCAG |
| PCR 1st Sense | 21,018S | 21,010–21,034 | GTACATACAGCTAATAAATGGGATC | |
| PCR 1st Antisense | 23,504AS | 23,496–23,518 | CCCTATTAAACAGCCTGCACGTG | |
| PCR 2nd Sense | 21,050S | 21,042–21,065 | TAGTGATATGTACGAC CCTAAGAC | |
| PCR 2nd Sense | 21,018S | 21,010–21,034 | GTACATACAGCTAATAAATGGGATC | |
| PCR 2nd Antisense | 23,461AS | 23,453–23,476 | TGTAGAATAAACACGCCAAGTAGG | |
| hCoV-full-S2 | PCR 1st Sense | 23,254S | 23,246–23,270 | TTYCAACAATTTGGCAGAGACATTG |
| PCR 1st Antisense | 25,666AS | 25,658–25,681 | CGAGCAAAAGGTGTGAGTAAACTG | |
| PCR 2nd Sense | 23,283S | 23,275–23,297 | CACTACTGATGCTGTCCGTGATC | |
| PCR 2nd Antisense | 25,619AS | 25,611–25,633 | AAACAAAGTGAACACCCTTGGAG |
| Primer Name | Nucleotide Sequence (5′-3′) | |
|---|---|---|
| hCoV-Spike-D | 22712-S | CCTACTAAATTAAATGATCTCTG |
| Spike-AS | CACCAGCAACTGTTTGTGGAC | |
| hCoV-full-a | 85-S | TTAAAATCTGTGTGGCTGTCACTCGG |
| a-2-2 | CGGGAGAAAATTGATCGTACAACAC | |
| a-1-Seq | TTAGGCGACGAGCTTGGCACTG | |
| hCoV-full-b | b-2-1 | AAACAAATTGTTGAATCCTGTGG |
| b-2-2 | CAACTTGCTTTTCACTCTTCATTTCC | |
| b-1-Seq | GTTAAATCCAGAGAAGAAACTGG | |
| b-S-1 | ATTGGCTTCACATATGTATTG | |
| b-AS-1 | CTTCAATAGTCTGAACAACTG | |
| hCoV-full-c | c-2-1 | CAGCACGAAGTTCTACTTGCACCA |
| c-2-2 | TTATAGTGACCACACTGGTAATTACC | |
| c-1-Seq | TGGAATTTGCGAGAAATGCTTG | |
| c-S-1 | AACAACTGTAGCGTCACTTATC | |
| hCoV-full-d | d-2-1 | GTGTTCAGATACCTTGTACGTGTGG |
| d-2-2 | CTGTAACACTATCAACGATGTAAGAAG | |
| d-1-Seq | ATTGTTTGGCATGTTAACAATG | |
| d-S1 | CTTCAAGAGAGCTTAAAGTTAC | |
| d-AS-1 | TAAGCCAAAAGCAGTTAAATCC | |
| hCoV-full-e | e-2-1 | TGTCTATGCTAATGGAGGTAAAGGC |
| e-2-2 | GCTTTGTGGAAATACAAATGATATAAGC | |
| e-S-1 | AATGTGTCCTTAGACAATGTC | |
| e-AS-1 | TGATCTTTCACAAGTGCCGTG | |
| hCoV-full-f | f-2-1 | ACTCATTCTTACCTGGTGTTTATTCTG |
| f-2-2 | ACAAAAGAGGCCAAAGTAACAAGTAC | |
| f-1-Seq | GAAGATTTACTCATTCGTAAGTC | |
| f-S-1 | AGTAACTTGTGGTACAACTAC | |
| f-AS-1 | TACGCATCACCCAACTAGCAG | |
| hCoV-full-g | g-2-1 | CTACTCAGGTGTAGTTACAACTGTC |
| g-2-2 | TAGCAAAACCAGCTACTTTATCATTG | |
| g-1-Seq | AGCAGGCTGTTGCTAATGGTG | |
| g-S1 | TTTCCATGCAGGGTGCTGTAG | |
| g-AS-1 | AATTGGCAGGCACTTCTGTTG | |
| hCoV-full-h | h-2-1 | CAACTCCGCGAACCCATGCTTCAG |
| h-2-2 | AAAAGTGCATTAACATTGGCCGTGAC | |
| h-1-Seq | TCAAGATCTCAATGGTAACTGG | |
| hS1 | ATTGTTGGTGTACTGACATTAG | |
| hAS1 | CTTGATCCTCATAACTCATTG | |
| hCoV-full-i | i-2-1 | GCTTGTCACACCGTTTCTATAGATTAG |
| i-2-2 | CTTAGTTAGCAATGTGCGTGGTGCAG | |
| i-1-Seq | GTCTTTAGCTATAGATGCTTACC | |
| i-S-1 | CAGATGGTACACTTATGATTG | |
| i-AS-1 | ATAGTGCTCTTGTGGCACTAG | |
| hCoV-full-j | j-2-1 | AATGCATTGCCTGAGACGACAGCAG |
| j-2-2 | ATTGCAACGTGTTATACACGTAGCAG | |
| j-1-Seq | GAAATTCCACGTAGGAATGTGG | |
| j-S-1 | TTGATTCATCACAGGGCTCAG | |
| j-AS-1 | CAGTTCATCACCAATTATAGG | |
| hCoV-full-k | k-2-1 | TGGTTGTGATGGTGGCAGTTTGTATG |
| k-2-2 | GGGGAAATTTACTCATGTCAAATAAAG | |
| k-1-Seq | TTCTATGACTGACATAGCCAAG | |
| k-S-1 | GCAACATTAAACCAGTACCAG | |
| k-AS-1 | CAACTCCTTTATCAGAACCAG | |
| hCoV-full-l | l-2-1 | TTTGTTTATGAGAATCTTCACAATTGG |
| l-2-2 | ATCAATCTCCATTGGTTGCTCTTCATC | |
| l-1-Seq | CTCAATTGAGTACAGACACTGG | |
| hCoV-full-m | m-2-1 | CTCGTTGACTTTCAGGTTACTATAGC |
| m-2-2 | TTGAGGAAGTTGTAGCACGATTGCAGC | |
| m-1-Seq | GGTTCTCACTTGAACTGCAAG | |
| hCoV-full-n | n-2-1 | ATGGTATTTCTACTACCTAGGAACTGG |
| n-2-2 | ATTAAAATCACATGGGGATAGCACTAC | |
| hCoV-full-S1 | 21018S | GTACATACAGCTAATAAATGGGATC |
| 23461AS | TGTAGAATAAACACGCCAAGTAGG | |
| S1-1-Seq | CACTGACACCACCAAAGAACAT | |
| S1-S1 | GTTAACAACTAAACGAACAATG | |
| S1-S2 | GTTCAGAGTTTATTCTAGTGCG | |
| S1-S3 | GGTGCTGCAGCTTATTATGTGG | |
| hCoV-full-S2 | 23283S | CACTACTGATGCTGTCCGTGATC |
| 25619AS | AAACAAAGTGAACACCCTTGGAG | |
| S2-2-Seq | AACGGCCTTACTGTTTGCCACC | |
| S2-S1 | ATTCAACTGAATGCAGCAATC | |
| S2-S2 | AACTTAGCTCCAAATTTGGTGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, K.; Takahashi, K.; Nagashima, S.; E, B.; Ouoba, S.; Takafuta, T.; Fujii, Y.; Mimori, M.; Okada, F.; Kishita, E.; et al. Exercising the Sanger Sequencing Strategy for Variants Screening and Full-Length Genome of SARS-CoV-2 Virus during Alpha, Delta, and Omicron Outbreaks in Hiroshima. Viruses 2022, 14, 720. https://doi.org/10.3390/v14040720
Ko K, Takahashi K, Nagashima S, E B, Ouoba S, Takafuta T, Fujii Y, Mimori M, Okada F, Kishita E, et al. Exercising the Sanger Sequencing Strategy for Variants Screening and Full-Length Genome of SARS-CoV-2 Virus during Alpha, Delta, and Omicron Outbreaks in Hiroshima. Viruses. 2022; 14(4):720. https://doi.org/10.3390/v14040720
Chicago/Turabian StyleKo, Ko, Kazuaki Takahashi, Shintaro Nagashima, Bunthen E, Serge Ouoba, Toshiro Takafuta, Yoshiki Fujii, Michi Mimori, Fumie Okada, Eisaku Kishita, and et al. 2022. "Exercising the Sanger Sequencing Strategy for Variants Screening and Full-Length Genome of SARS-CoV-2 Virus during Alpha, Delta, and Omicron Outbreaks in Hiroshima" Viruses 14, no. 4: 720. https://doi.org/10.3390/v14040720
APA StyleKo, K., Takahashi, K., Nagashima, S., E, B., Ouoba, S., Takafuta, T., Fujii, Y., Mimori, M., Okada, F., Kishita, E., Ariyoshi, K., Hussain, M. R. A., Sugiyama, A., Akita, T., Kuwabara, M., & Tanaka, J. (2022). Exercising the Sanger Sequencing Strategy for Variants Screening and Full-Length Genome of SARS-CoV-2 Virus during Alpha, Delta, and Omicron Outbreaks in Hiroshima. Viruses, 14(4), 720. https://doi.org/10.3390/v14040720