
Mapping the Key Residues within the Porcine Reproductive and Respiratory Syndrome Virus nsp1α Replicase Protein Required for Degradation of Swine Leukocyte Antigen Class I Molecules

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Infection
2.2. Enzymes, Antibodies, and Chemicals
2.3. Plasmid Construction
2.4. Site-Directed Mutagenesis of PRRSV JXwn06 nsp1α and Virus Rescue
2.5. Growth Properties of Viral Mutants
2.6. Confocal Microscopy
2.7. Flow Cytometry Analysis
2.8. Immunoprecipitation and Ubiquitination Assays
2.9. Western Blot Analysis
2.10. Statistical Analysis
3. Results
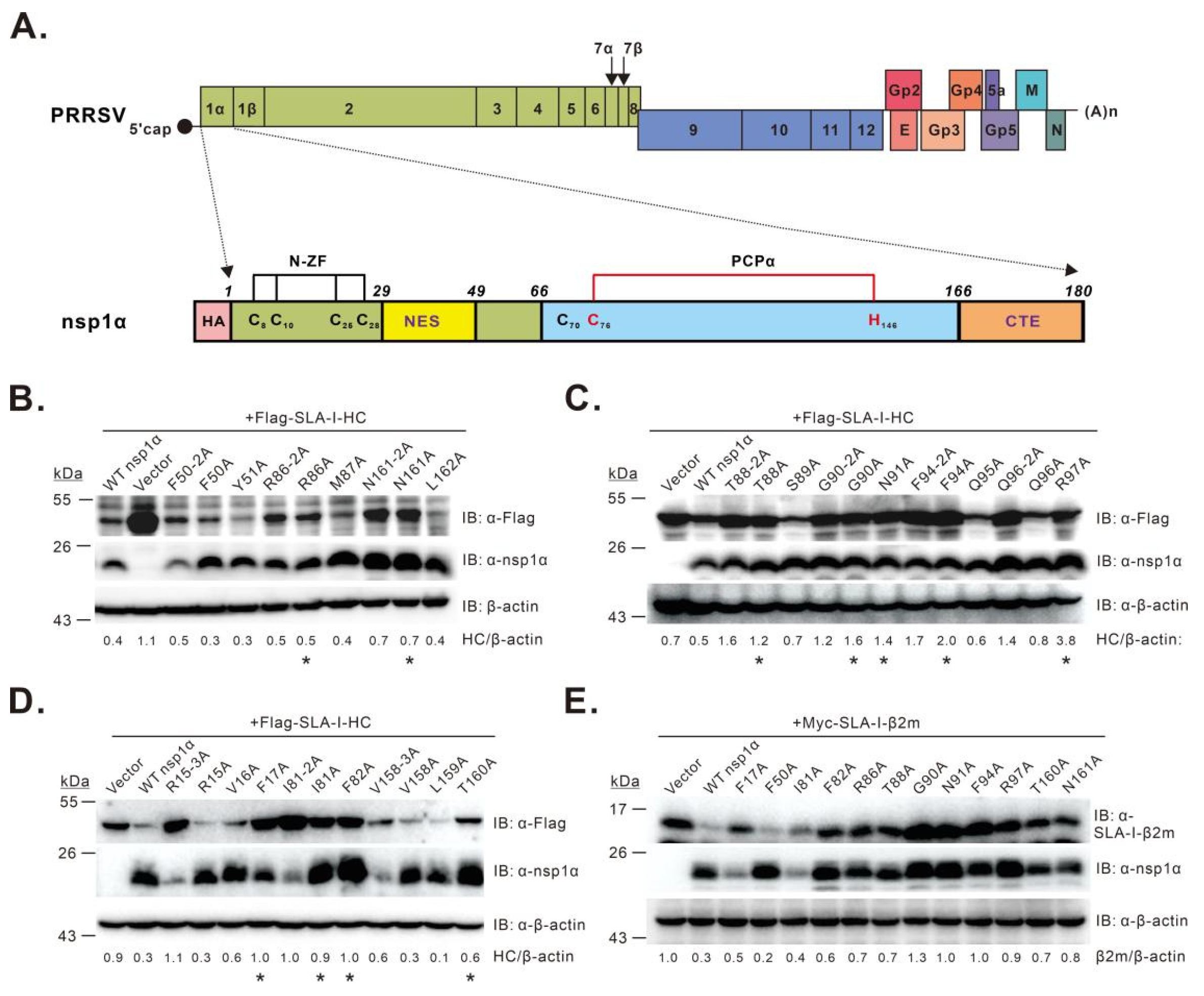
3.1. Screening of nsp1α Residues Critical for Inducing SLA-I Degradation
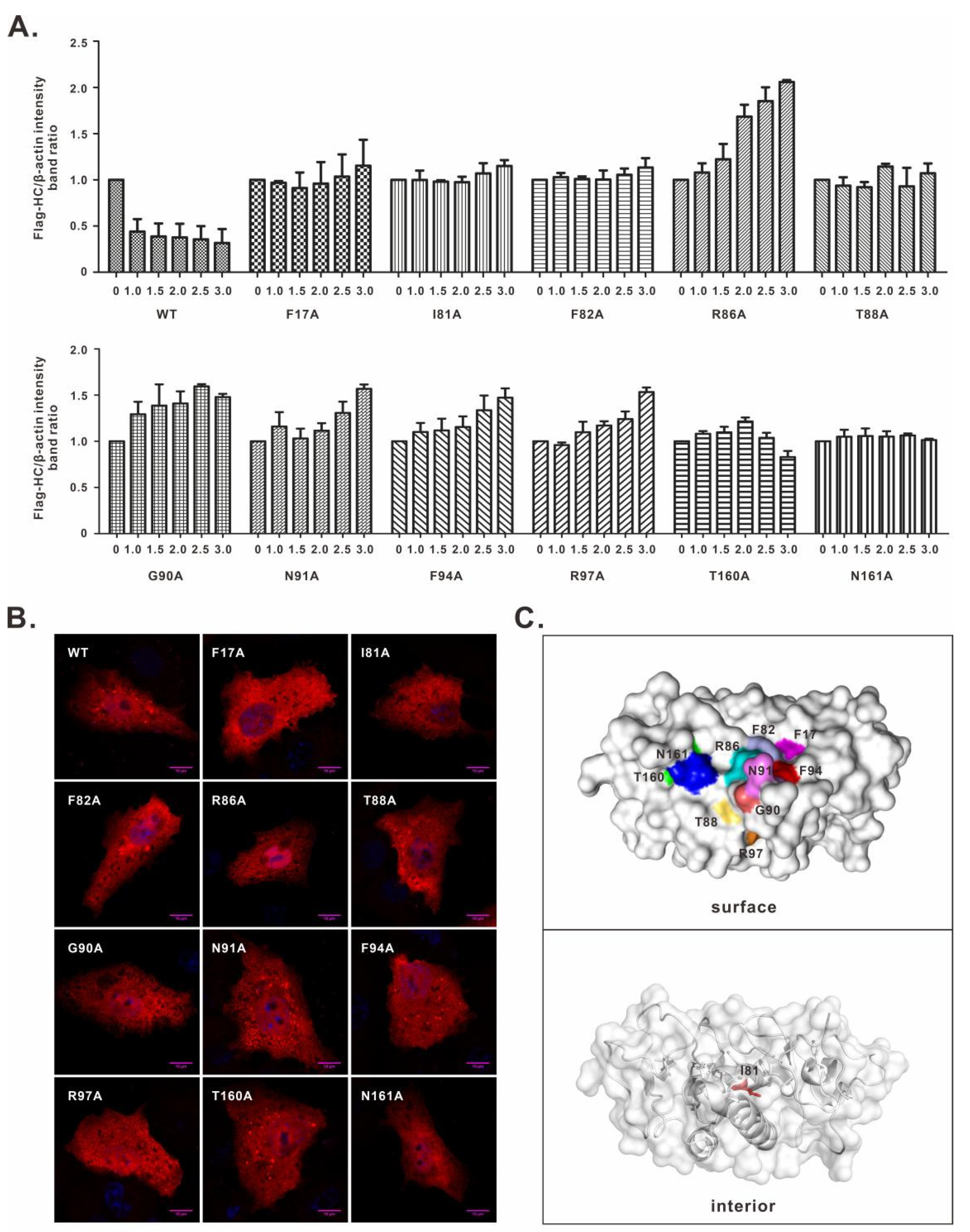
3.2. Characterization of the nsp1α Mutants with Decreased Degradation Activity
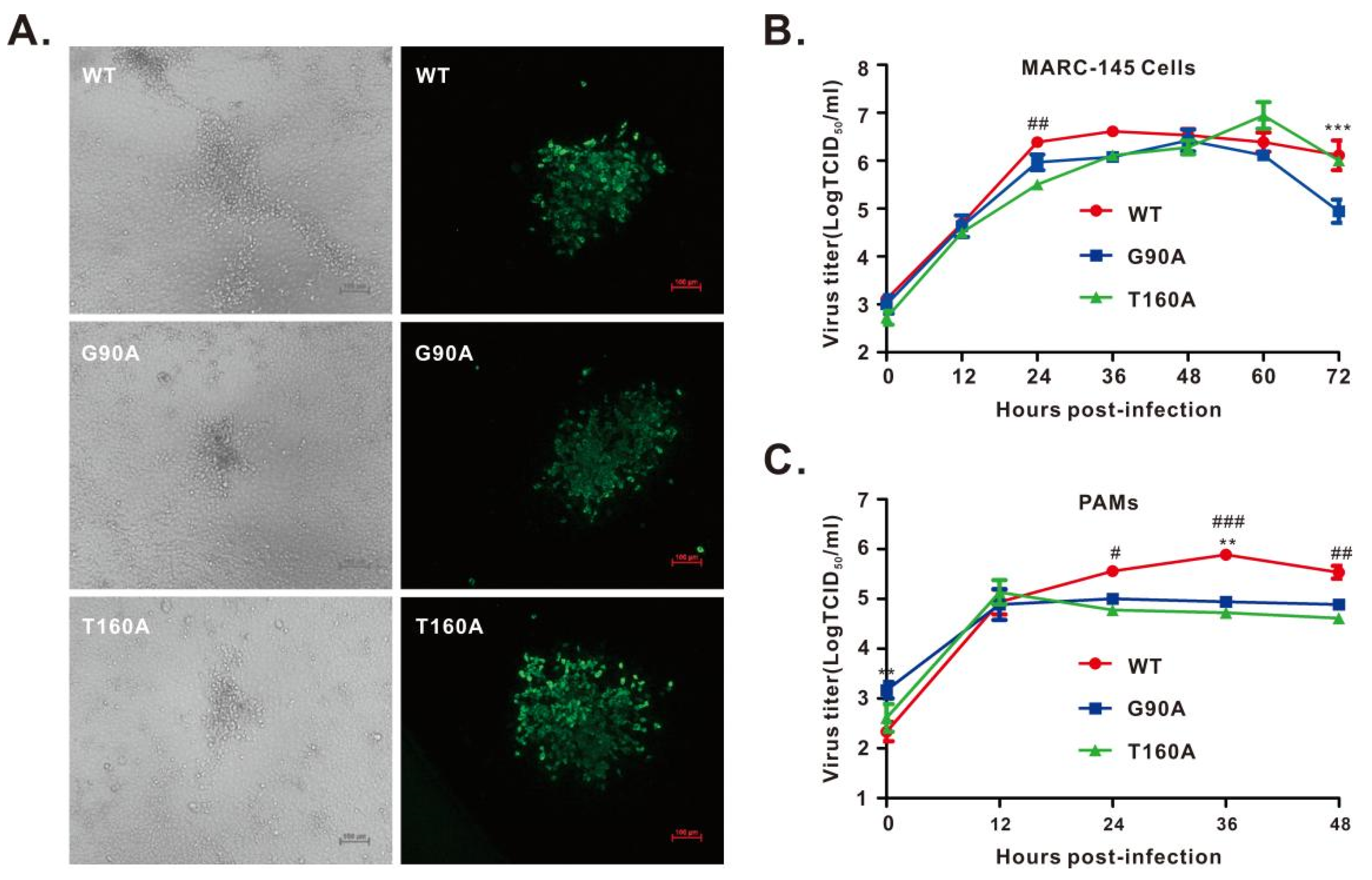
3.3. Recovery and Growth Kinetics of nsp1α Mutant Viruses
3.4. Co-Localization Analysis of nsp1α Mutants with SLA-I in PRRSV-Infected PAMs
3.5. PRRSV Strain JXwn06 Carrying the G90A Mutation in nsp1α Failed to Downregulate SLA-I in PAMs
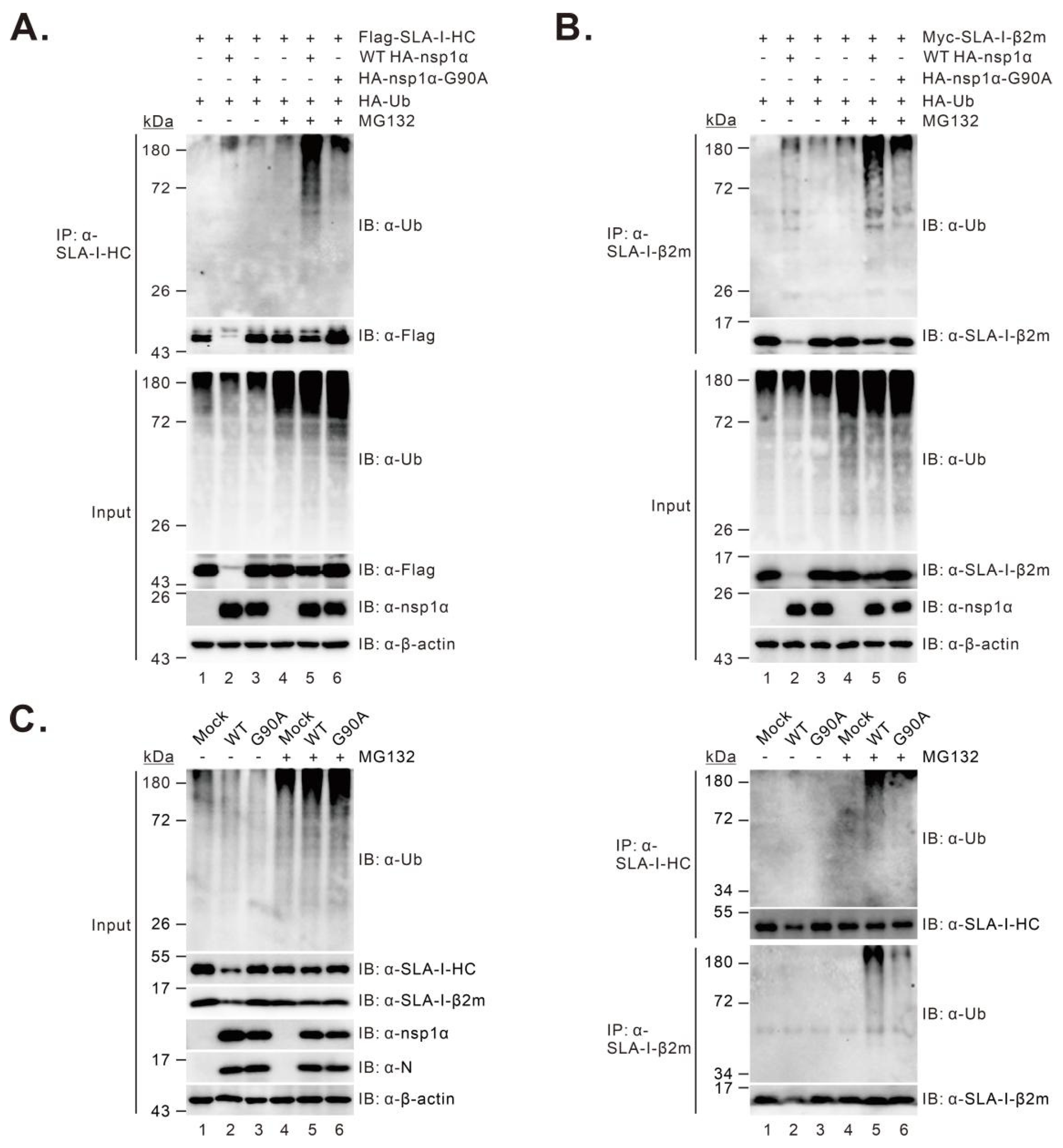
3.6. The G90A Mutation of nsp1α Results in Decreased SLA-I Ubiquitination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Han, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2017, 209, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.X.; Pattnaik, A.K.; Osorio, F.A. Strategies to broaden the cross-protective efficacy of vaccines against porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2017, 206, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Lunney, J.K.; Benfield, D.A.; Rowland, R.R. Porcine reproductive and respiratory syndrome virus: An update on an emerging and re-emerging viral disease of swine. Virus Res. 2010, 154, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wensvoort, G.; Terpstra, C.; Pol, J.M.A.; Terlaak, E.A.; Bloemraad, M.; Dekluyver, E.P.; Kragten, C.; Vanbuiten, L.; Denbesten, A.; Wagenaar, F.; et al. Mystery Swine Disease in the Netherlands-the Isolation of Lelystad Virus. Vet. Q. 1991, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Dea, S.; Bilodeau, R.; Athanassious, R.; Sauvageau, R.A.; Martineau, G.P. Isolation of the Porcine Reproductive and Respiratory Syndrome Virus in Quebec. Can. Vet. J. Rev. Vet. Can. 1992, 33, 552–553. [Google Scholar]
- Zhou, L.; Ge, X.; Yang, H. Porcine Reproductive and Respiratory Syndrome Modified Live Virus Vaccine: A "Leaky" Vaccine with Debatable Efficacy and Safety. Vaccines 2021, 9, 362. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, C. Porcine reproductive and respiratory syndrome virus vaccines: Current status and strategies to a universal vaccine. Transbound. Emerg. Dis. 2014, 61, 109–120. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Gu, G.; Sun, W.; Zhang, Y.J.; Zhou, E.M. Improved Vaccine against PRRSV: Current Progress and Future Perspective. Front. Microbiol. 2017, 8, 1635. [Google Scholar] [CrossRef]
- Renukaradhya, G.J.; Meng, X.J.; Calvert, J.G.; Roof, M.; Lager, K.M. Live porcine reproductive and respiratory syndrome virus vaccines: Current status and future direction. Vaccine 2015, 33, 4069–4080. [Google Scholar] [CrossRef]
- Wang, H.; Xu, Y.; Feng, W. Porcine Reproductive and Respiratory Syndrome Virus: Immune Escape and Application of Reverse Genetics in Attenuated Live Vaccine Development. Vaccines 2021, 9, 480. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Renukaradhya, G.J.; Alekseev, K.P.; Fang, Y.; Tang, Y.; Saif, L.J. Porcine reproductive and respiratory syndrome virus modifies innate immunity and alters disease outcome in pigs subsequently infected with porcine respiratory coronavirus: Implications for respiratory viral co-infections. J. Gen. Virol. 2009, 90, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Xiao, S.; Jiang, Y.; Jin, H.; Wang, D.; Liu, M.; Chen, H.; Fang, L. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol. Immunol. 2008, 45, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Nicolas, O.; Quereda, J.J.; Gomez-Laguna, J.; Javier Salguero, F.; Carrasco, L.; Ramis, G.; Jose Pallares, F. Cytokines transcript levels in lung and lymphoid organs during genotype 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) infection. Vet. Immunol. Immunopathol. 2014, 160, 26–40. [Google Scholar] [CrossRef]
- Murtaugh, M.P.; Xiao, Z.G.; Zuckermann, F. Immunological responses of swine to porcine reproductive and respiratory syndrome virus infection. Viral Immunol. 2002, 15, 533–547. [Google Scholar] [CrossRef]
- Loemba, H.D.; Mounir, S.; Mardassi, H.; Archambault, D.; Dea, S. Kinetics of humoral immune response to the major structural proteins of the porcine reproductive and respiratory syndrome virus. Arch. Virol. 1996, 141, 751–761. [Google Scholar] [CrossRef]
- Meier, W.A.; Wheeler, J.; Husmann, R.J.; Osorio, F.A.; Zuckermann, F.A.; Aasp, A. Characteristics of the immune response of pigs to wild-type PRRS virus or to commercially available vaccines: An unconventional response. In Proceedings of the 2000 American Association of Swine Practitioners Annual Meeting, Indianapolis, Indiana, 11–14 March 2000; pp. 415–418. [Google Scholar]
- Loving, C.L.; Osorio, F.A.; Murtaugh, M.P.; Zuckermann, F.A. Innate and adaptive immunity against Porcine Reproductive and Respiratory Syndrome Virus. Vet. Immunol. Immunopathol. 2015, 167, 1–14. [Google Scholar] [CrossRef]
- Xiao, Z.; Batista, L.; Dee, S.; Halbur, P.; Murtaugh, M.P. The level of virus-specific T-cell and macrophage recruitment in porcine reproductive and respiratory syndrome virus infection in pigs is independent of virus load. J. Virol. 2004, 78, 5923–5933. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, J.-L.; Cheng, Y.; Wang, J.-X.; Zou, Z. Pattern recognition receptors from lepidopteran insects and their biological functions. Dev. Comp. Immunol. 2020, 108, 103688. [Google Scholar] [CrossRef]
- Fritzlar, S.; Jegaskanda, S.; Aktepe, T.E.; Prier, J.E.; Holz, L.E.; White, P.A.; Mackenzie, J.M. Mouse Norovirus Infection Reduces the Surface Expression of Major Histocompatibility Complex Class I Proteins and Inhibits CD8(+) T Cell Recognition and Activation. J. Virol. 2018, 92, e00286-18. [Google Scholar] [CrossRef] [PubMed]
- Wodarz, D. The persistence of CTL memory. Neth. J. Med. 2002, 60, 4–13; discussion 14–16. [Google Scholar] [PubMed]
- Kutsch, O.; Vey, T.; Kerkau, T.; Hunig, T.; Schimpl, A. HIV type 1 abrogates TAP-mediated transport of antigenic peptides presented by MHC class I. Transporter associated with antigen presentation. AIDS Res. Hum. Retrovir. 2002, 18, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, G.B.; Mahmutefendic, H.; Tomas, M.I.; Kucic, N.; Le Bouteiller, P.; Lucin, P. Early endosomal rerouting of major histocompatibility class I conformers. J. Cell Physiol. 2012, 227, 2953–2964. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, H.S.; Seo, S.H. Characterization of interaction between porcine reproductive and respiratory syndrome virus and porcine dendritic cells. J. Microbiol. Biotechnol. 2008, 18, 1709–1716. [Google Scholar]
- Wang, X.; Eaton, M.; Mayer, M.; Li, H.; He, D.; Nelson, E.; Christopher-Hennings, J. Porcine reproductive and respiratory syndrome virus productively infects monocyte-derived dendritic cells and compromises their antigen-presenting ability. Arch. Virol. 2007, 152, 289–303. [Google Scholar] [CrossRef]
- Du, J.; Ge, X.; Liu, Y.; Jiang, P.; Wang, Z.; Zhang, R.; Zhou, L.; Guo, X.; Han, J.; Yang, H. Targeting Swine Leukocyte Antigen Class I Molecules for Proteasomal Degradation by the nsp1alpha Replicase Protein of the Chinese Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus Strain JXwn06. J. Virol. 2016, 90, 682–693. [Google Scholar] [CrossRef]
- Cao, Q.M.; Subramaniam, S.; Ni, Y.Y.; Cao, D.; Meng, X.J. The non-structural protein Nsp2TF of porcine reproductive and respiratory syndrome virus down-regulates the expression of Swine Leukocyte Antigen class I. Virology 2016, 491, 115–124. [Google Scholar] [CrossRef]
- Qi, P.; Liu, K.; Wei, J.; Li, Y.; Li, B.; Shao, D.; Wu, Z.; Shi, Y.; Tong, G.; Qiu, Y.; et al. Nonstructural Protein 4 of Porcine Reproductive and Respiratory Syndrome Virus Modulates Cell Surface Swine Leukocyte Antigen Class I Expression by Downregulating beta2-Microglobulin Transcription. J. Virol. 2017, 91, e01755-16. [Google Scholar] [CrossRef]
- Sun, Y.; Xue, F.; Guo, Y.; Ma, M.; Hao, N.; Zhang, X.C.; Lou, Z.; Li, X.; Rao, Z. Crystal Structure of Porcine Reproductive and Respiratory Syndrome Virus Leader Protease Nsp1 alpha. J. Virol. 2009, 83, 10931–10940. [Google Scholar] [CrossRef]
- Kappes, M.A.; Faaberg, K.S. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guo, X.; Ge, X.; Chen, Y.; Sun, Q.; Yang, H. Changes in the cellular proteins of pulmonary alveolar macrophage infected with porcine reproductive and respiratory syndrome virus by proteomics analysis. J. Proteome Res. 2009, 8, 3091–3097. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, J.; Zeng, J.; Yin, S.; Li, Y.; Zheng, L.; Guo, X.; Ge, X.; Yang, H. The 30-amino-acid deletion in the Nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China is not related to its virulence. J. Virol. 2009, 83, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, J.; Ge, X.; Zhou, L.; Guo, X.; Yang, H. Prokaryotic expression of SLA-I alpha chain and beta chain and antiserum preparation. Acta Vet. Et Zootech. Sin. 2015, 46, 1224–1231. [Google Scholar]
- Song, J.; Gao, P.; Kong, C.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. The nsp2 Hypervariable Region of Porcine Reproductive and Respiratory Syndrome Virus Strain JXwn06 Is Associated with Viral Cellular Tropism to Primary Porcine Alveolar Macrophages. J. Virol. 2019, 93, e01436-19. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, L.; Zhang, J.; Ge, X.; Zhou, R.; Zheng, H.; Geng, G.; Guo, X.; Yang, H. Nsp9 and Nsp10 contribute to the fatal virulence of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China. PLoS Pathog. 2014, 10, e1004216. [Google Scholar] [CrossRef]
- Woolard, S.N.; Kumaraguru, U. Viral vaccines and CTL response. J. Biomed. Biotechnol. 2010, 2010, 141657. [Google Scholar] [CrossRef]
- den Boon, J.A.; Faaberg, K.S.; Meulenberg, J.J.; Wassenaar, A.L.; Plagemann, P.G.; Gorbalenya, A.E.; Snijder, E.J. Processing and evolution of the N-terminal region of the arterivirus replicase ORF1a protein: Identification of two papainlike cysteine proteases. J. Virol. 1995, 69, 4500–4505. [Google Scholar] [CrossRef]
- Chen, Z.; Lawson, S.; Sun, Z.; Zhou, X.; Guan, X.; Christopher-Hennings, J.; Nelson, E.A.; Fang, Y. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: Nsp1 function as interferon antagonist. Virology 2010, 398, 87–97. [Google Scholar] [CrossRef]
- Kroese, M.V.; Zevenhoven-Dobbe, J.C.; Bos-de Ruijter, J.N.A.; Peeters, B.P.H.; Meulenberg, J.J.M.; Cornelissen, L.; Snijder, E.J. The nsp1alpha and nsp1 papain-like autoproteinases are essential for porcine reproductive and respiratory syndrome virus RNA synthesis. J. Gen. Virol. 2008, 89, 494–499. [Google Scholar] [CrossRef]
- Shi, X.B.; Zhang, X.Z.; Wang, F.Y.; Wang, L.; Qiao, S.L.; Guo, J.Q.; Luo, C.H.; Wan, B.; Deng, R.G.; Zhang, G.P. The Zinc-Finger Domain Was Essential for Porcine Reproductive and Respiratory Syndrome Virus Nonstructural Protein-1 alpha to Inhibit the Production of Interferon-beta. J. Interferon Cytokine Res. 2013, 33, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, G.; Wang, L.; Li, X.; Zhi, Y.; Wang, F.; Fan, J.; Deng, R. The nonstructural protein 1 papain-like cysteine protease was necessary for porcine reproductive and respiratory syndrome virus nonstructural protein 1 to inhibit interferon-beta induction. DNA Cell Biol. 2011, 30, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Su, Y.; Li, R.; Zhang, L.; Chen, C.; Zhang, L.; Faaberg, K.; Huang, J. Dual Regulation of Host TRAIP Post-translation and Nuclear/Plasma Distribution by Porcine Reproductive and Respiratory Syndrome Virus Non-structural Protein 1alpha Promotes Viral Proliferation. Front. Immunol. 2018, 9, 3023. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, S.; Sun, W.; Chen, L.; Yoo, D.; Li, F.; Ren, S.; Guo, L.; Cong, X.; Li, J.; et al. Nuclear export signal of PRRSV NSP1alpha is necessary for type I IFN inhibition. Virology 2016, 499, 278–287. [Google Scholar] [CrossRef]
- Beura, L.K.; Subramaniam, S.; Vu, H.L.; Kwon, B.; Pattnaik, A.K.; Osorio, F.A. Identification of amino acid residues important for anti-IFN activity of porcine reproductive and respiratory syndrome virus non-structural protein 1. Virology 2012, 433, 431–439. [Google Scholar] [CrossRef][Green Version]
- Han, M.; Du, Y.; Song, C.; Yoo, D. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1alpha subunit. Virus Res. 2013, 172, 54–65. [Google Scholar] [CrossRef]
- Song, C.; Krell, P.; Yoo, D. Nonstructural protein 1alpha subunit-based inhibition of NF-kappaB activation and suppression of interferon-beta production by porcine reproductive and respiratory syndrome virus. Virology 2010, 407, 268–280. [Google Scholar] [CrossRef]
- Subramaniam, S.; Kwon, B.; Beura, L.K.; Kuszynski, C.A.; Pattnaik, A.K.; Osorio, F.A. Porcine reproductive and respiratory syndrome virus non-structural protein 1 suppresses tumor necrosis factor-alpha promoter activation by inhibiting NF-kappaB and Sp1. Virology 2010, 406, 270–279. [Google Scholar] [CrossRef]
- Park, I.B.; Choi, Y.C.; Lee, K.T.; Chun, T. Transcriptome analysis of pig macrophages expressing porcine reproductive and respiratory syndrome virus non-structural protein 1. Vet. Immunol. Immunopathol. 2021, 231, 110147. [Google Scholar] [CrossRef]
- Chen, X.; Bai, J.; Liu, X.; Song, Z.; Zhang, Q.; Wang, X.; Jiang, P. Nsp1alpha of Porcine Reproductive and Respiratory Syndrome Virus Strain BB0907 Impairs the Function of Monocyte-Derived Dendritic Cells via the Release of Soluble CD83. J. Virol. 2018, 92, e00366-18. [Google Scholar] [CrossRef]
- Jing, H.; Fang, L.; Ding, Z.; Wang, D.; Hao, W.; Gao, L.; Ke, W.; Chen, H.; Xiao, S. Porcine Reproductive and Respiratory Syndrome Virus nsp1alpha Inhibits NF-kappaB Activation by Targeting the Linear Ubiquitin Chain Assembly Complex. J. Virol. 2017, 91, e01911-16. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.; Sun, Y.; Lai, F.W.; Song, C.; Yoo, D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology 2010, 402, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shi, X.; Zhang, X.; Wang, L.; Luo, J.; Xing, G.; Deng, R.; Yang, H.; Li, J.; Wang, A.; et al. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Inhibits RNA-Mediated Gene Silencing by Targeting Ago-2. Viruses 2015, 7, 5539–5552. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Beura, L.K.; Kwon, B.; Pattnaik, A.K.; Osorio, F.A. Amino acid residues in the non-structural protein 1 of porcine reproductive and respiratory syndrome virus involved in down-regulation of TNF-alpha expression in vitro and attenuation in vivo. Virology 2012, 432, 241–249. [Google Scholar] [CrossRef]
- Roeth, J.F.; Williams, M.; Kasper, M.R.; Filzen, T.M.; Collins, K.L. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 2004, 167, 903–913. [Google Scholar] [CrossRef]
- Cho, S.; Kim, B.Y.; Ahn, K.; Jun, Y. The C-terminal amino acid of the MHC-I heavy chain is critical for binding to Derlin-1 in human cytomegalovirus US11-induced MHC-I degradation. PLoS ONE 2013, 8, e72356. [Google Scholar] [CrossRef]
- Hill, A.; Jugovic, P.; York, I.; Russ, G.; Bennink, J.; Yewdell, J.; Ploegh, H.; Johnson, D. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995, 375, 411–415. [Google Scholar] [CrossRef]
- Wei, H.Y.; Wang, Y.; Chowdhury, S.I. Bovine Herpesvirus Type 1 (BHV-1) U(L)49.5 Luminal Domain Residues 30 to 32 Are Critical for MHC-I Down-Regulation in Virus-Infected Cells. PLoS ONE 2011, 6, e25742. [Google Scholar] [CrossRef]
- Mansouri, M.; Bartee, E.; Gouveia, K.; Nerenberg, B.T.H.; Barrett, J.; Thomas, L.; Thomas, G.; McFadden, G.; Fruh, K. The PHD/LAP-domain protein M153R of myxomavirus is a ubiquitin ligase that induces the rapid internalization and lysosomal destruction of CD4. J. Virol. 2003, 77, 1427–1440. [Google Scholar] [CrossRef]
- Zhu, S.; Regev, D.; Watanabe, M.; Hickman, D.; Moussatche, N.; Jesus, D.M.; Kahan, S.M.; Napthine, S.; Brierley, I.; Hunter, R.N., 3rd; et al. Identification of immune and viral correlates of norovirus protective immunity through comparative study of intra-cluster norovirus strains. PLoS Pathog. 2013, 9, e1003592. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Lencer, W.; Hansen, T.H. The viral E3 ubiquitin ligase mK3 uses the Derlin/p97 endoplasmic reticulum-associated degradation pathway to mediate down-regulation of major histocompatibility complex class I proteins. J. Biol. Chem. 2006, 281, 8636–8644. [Google Scholar] [CrossRef] [PubMed]
- Majumder, B.; Gray, B.; McBurney, S.; Schaefer, T.M.; Dentchev, T.; Mahalingam, S.; Reinhart, T.A.; Ayyavoo, V. Attenuated nef DNA vaccine construct induces cellular immune response: Role in HIV-1 multiprotein vaccine. Immunol. Lett. 2003, 89, 207–214. [Google Scholar] [CrossRef]
- Peng, R.; Voltan, R.; Cristillo, A.D.; Alvord, W.G.; Davis-Warren, A.; Zhou, Q.F.; Murthy, K.K.; Robert-Guroff, M. Replicating Ad-recombinants encoding non-myristoylated rather than wild-type HIV Nef elicit enhanced cellular immunity. AIDS 2006, 20, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; He, J.; Paulsen, D.B.; Chowdhury, S.I. Bovine herpesvirus type 1 (BHV-1) mutant lacking U(L)49.5 luminal domain residues 30-32 and cytoplasmic tail residues 80-96 induces more rapid onset of virus neutralizing antibody and cellular immune responses in calves than the wild-type strain Cooper. Vet. Immunol. Immunopathol. 2012, 147, 223–229. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Gao, P.; Zhou, L.; Ge, X.; Zhang, Y.; Guo, X.; Han, J.; Yang, H. Mapping the Key Residues within the Porcine Reproductive and Respiratory Syndrome Virus nsp1α Replicase Protein Required for Degradation of Swine Leukocyte Antigen Class I Molecules. Viruses 2022, 14, 690. https://doi.org/10.3390/v14040690
Liu Y, Gao P, Zhou L, Ge X, Zhang Y, Guo X, Han J, Yang H. Mapping the Key Residues within the Porcine Reproductive and Respiratory Syndrome Virus nsp1α Replicase Protein Required for Degradation of Swine Leukocyte Antigen Class I Molecules. Viruses. 2022; 14(4):690. https://doi.org/10.3390/v14040690
Chicago/Turabian StyleLiu, Yuanyuan, Peng Gao, Lei Zhou, Xinna Ge, Yongning Zhang, Xin Guo, Jun Han, and Hanchun Yang. 2022. "Mapping the Key Residues within the Porcine Reproductive and Respiratory Syndrome Virus nsp1α Replicase Protein Required for Degradation of Swine Leukocyte Antigen Class I Molecules" Viruses 14, no. 4: 690. https://doi.org/10.3390/v14040690
APA StyleLiu, Y., Gao, P., Zhou, L., Ge, X., Zhang, Y., Guo, X., Han, J., & Yang, H. (2022). Mapping the Key Residues within the Porcine Reproductive and Respiratory Syndrome Virus nsp1α Replicase Protein Required for Degradation of Swine Leukocyte Antigen Class I Molecules. Viruses, 14(4), 690. https://doi.org/10.3390/v14040690