
CovDif, a Tool to Visualize the Conservation between SARS-CoV-2 Genomes and Variants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
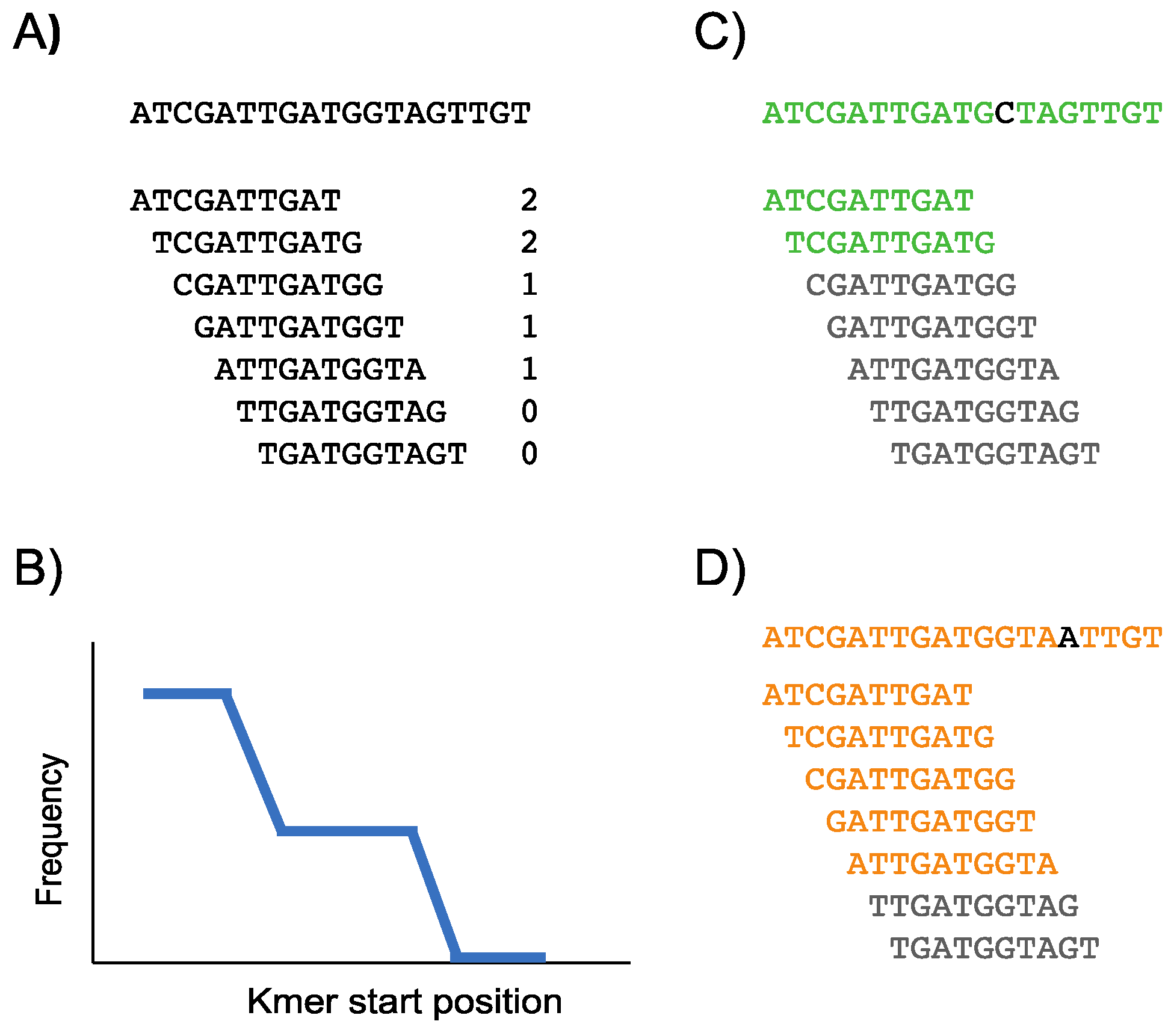
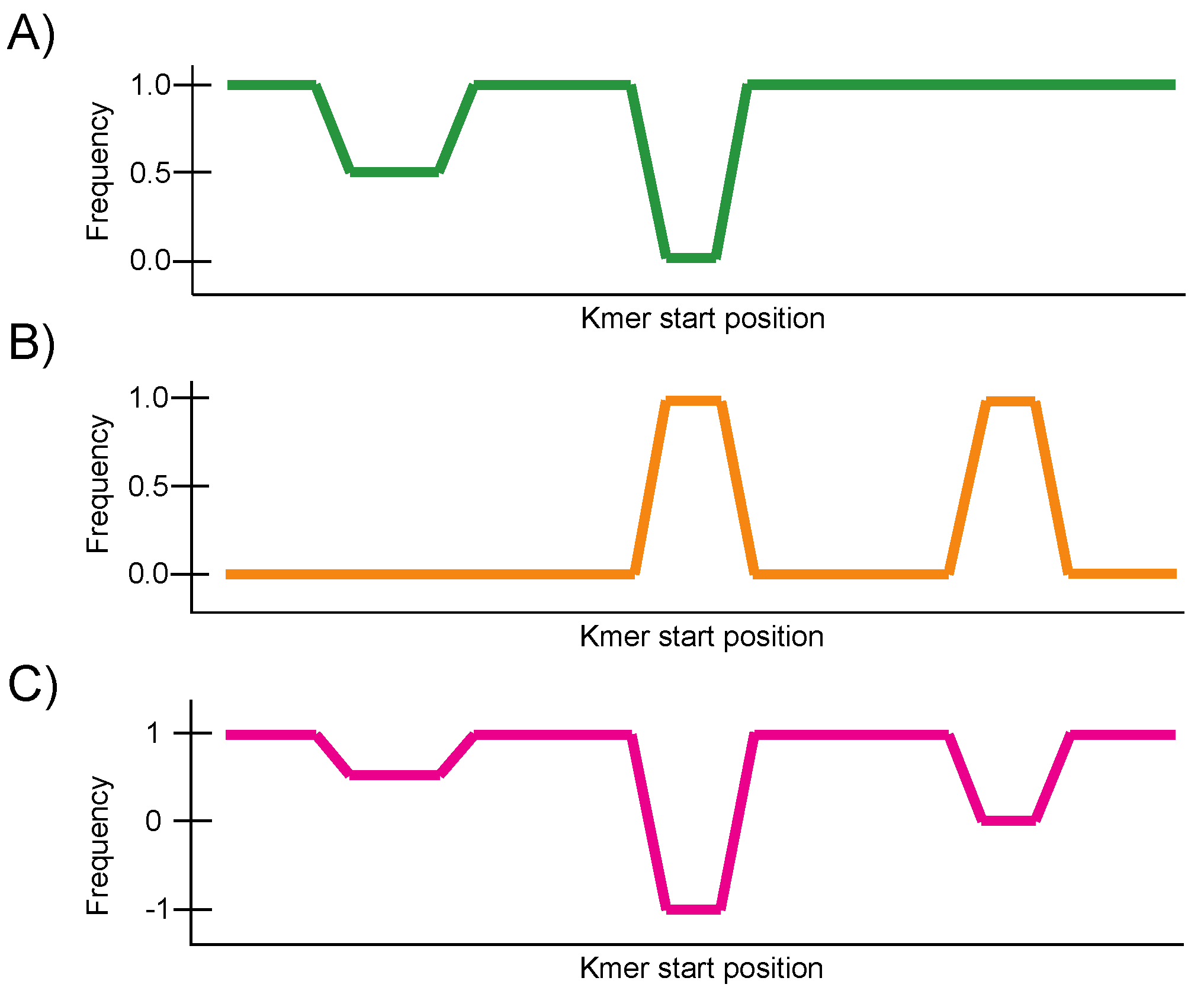
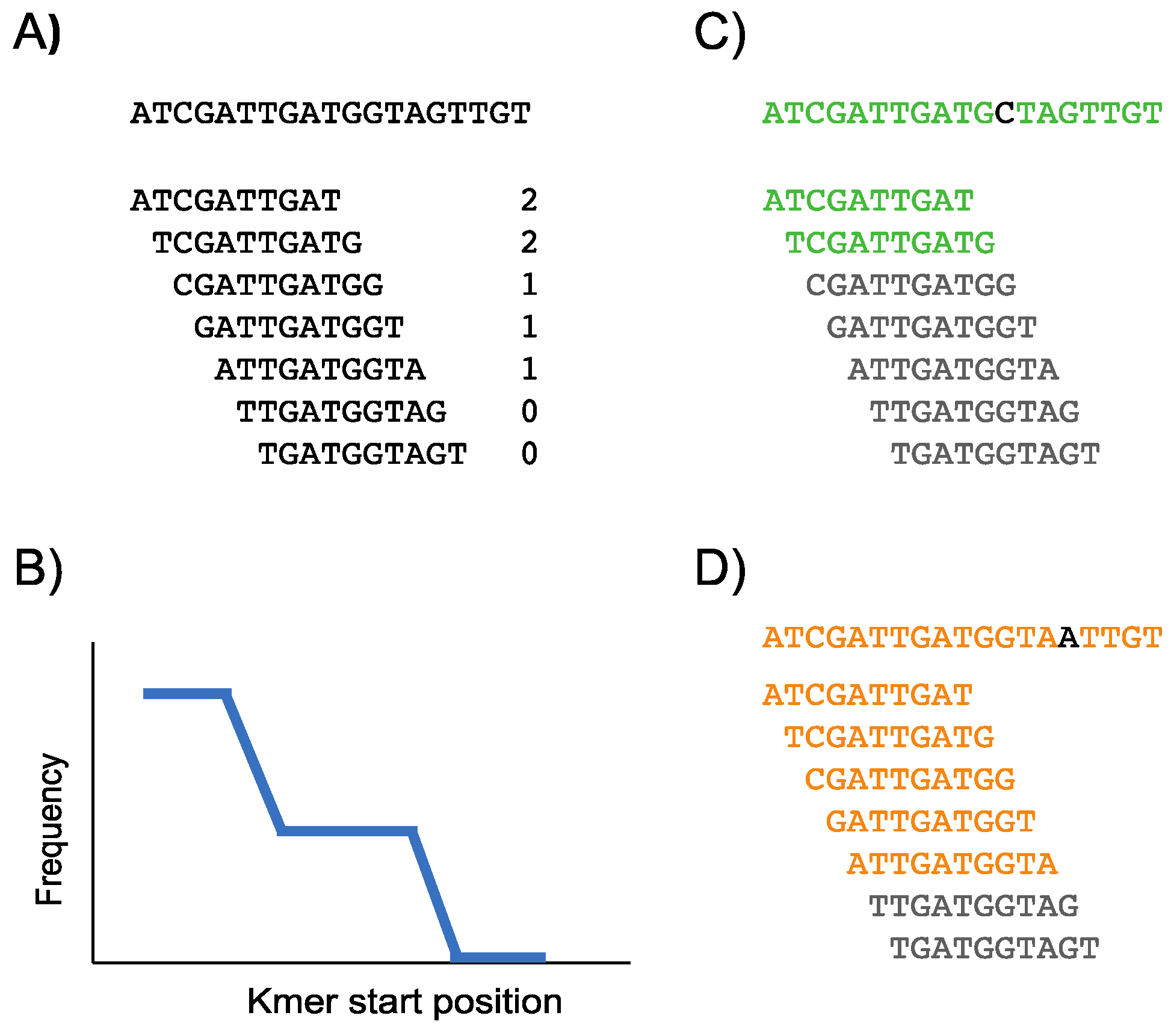
2.2. Construction of the Conservation Landscape
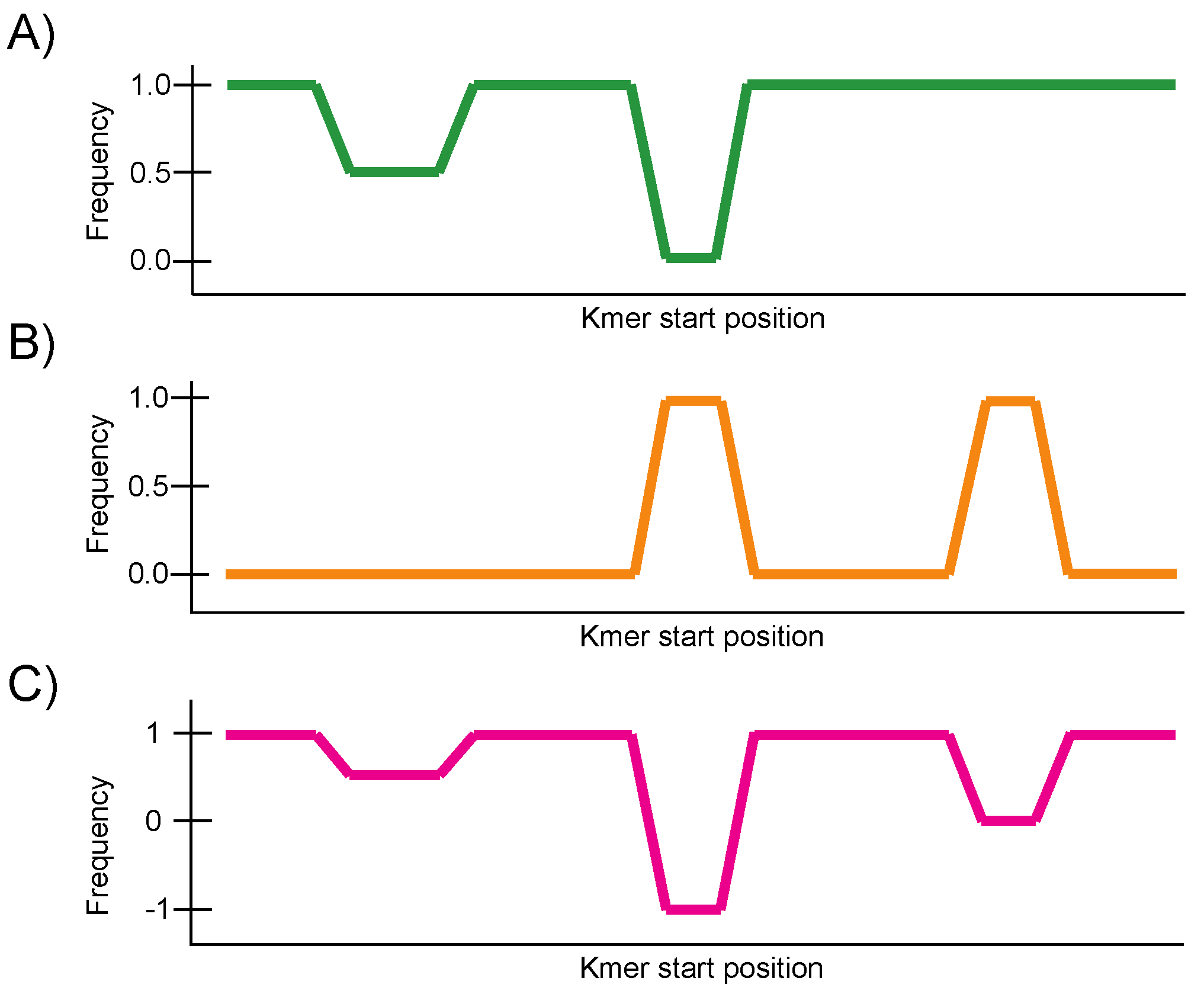
2.3. Construction of the Differential Landscape
2.4. Genome Browser Interface and Analysis of Conservation and Differential Landscapes
2.5. CovDif Implementation
2.6. Annotation of Known Variants
2.7. Obtainment of Primer and Probe Sequences from Diagnostic Kits
3. Results
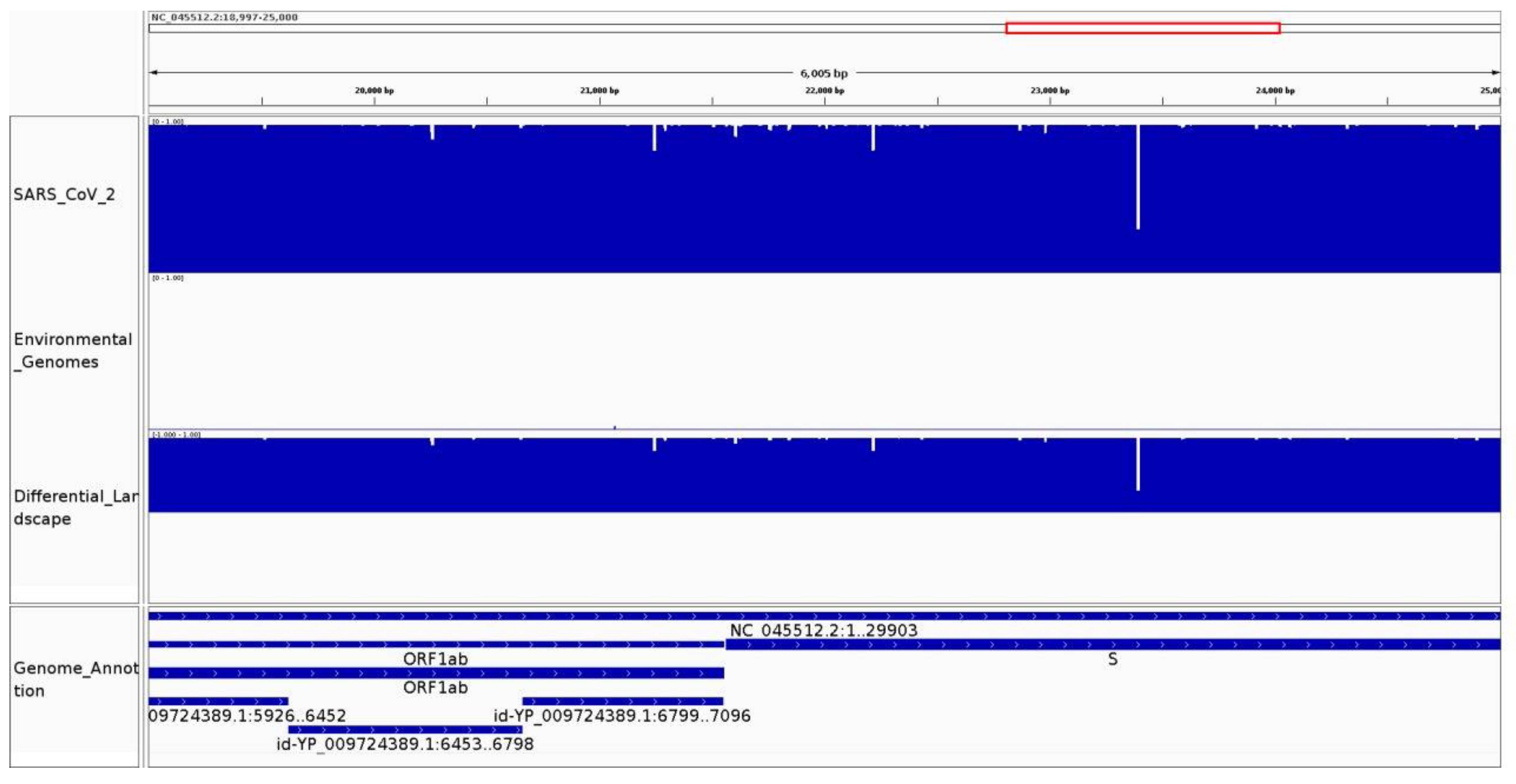
3.1. Genomic Variation Is Highlighted along the Conservation and Differential Landscapes
3.2. CovDif Can Be Used for a Wide-Range of Applications
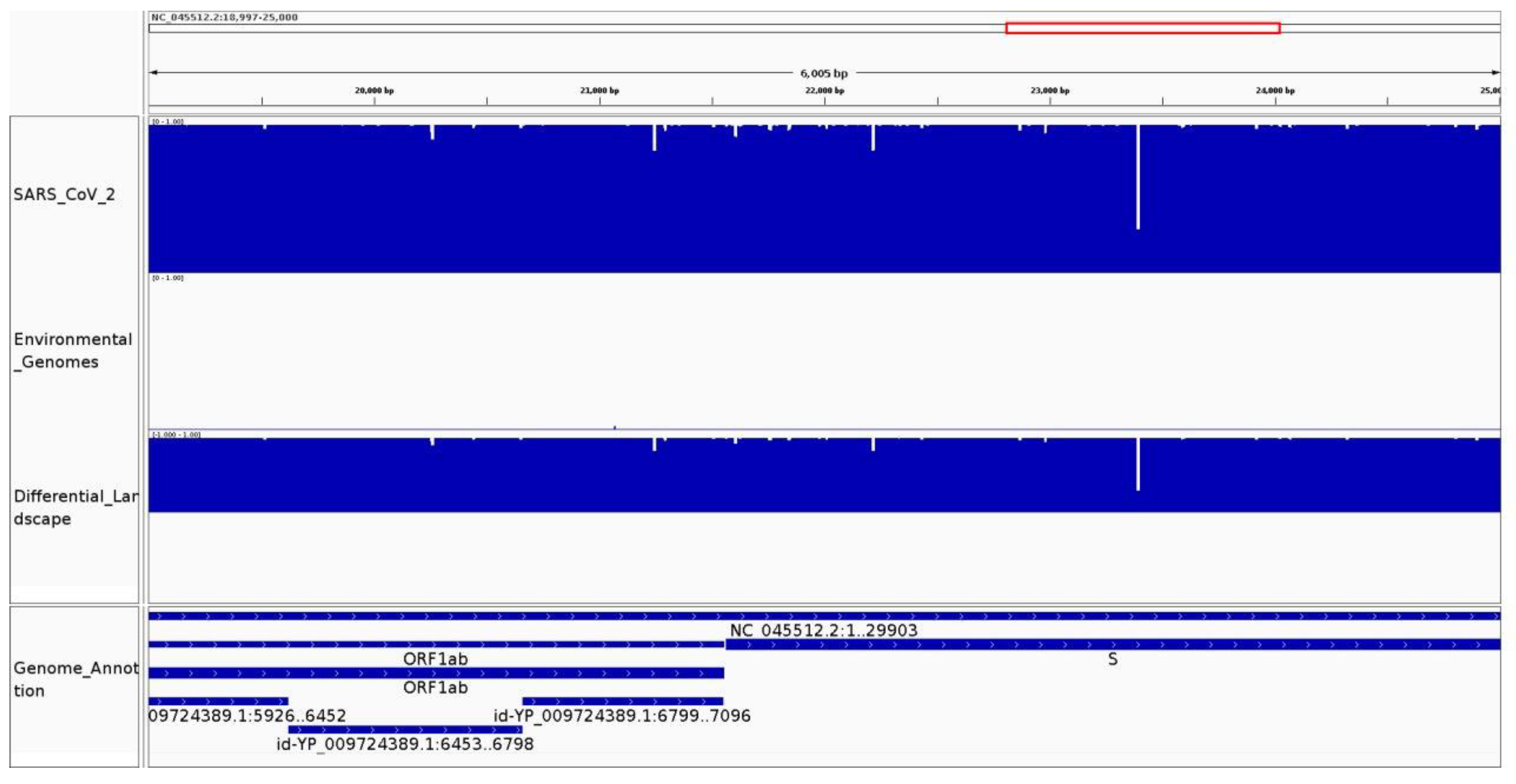
3.2.1. It Can Be Used to Identify Species-Specific and Highly Conserved Regions
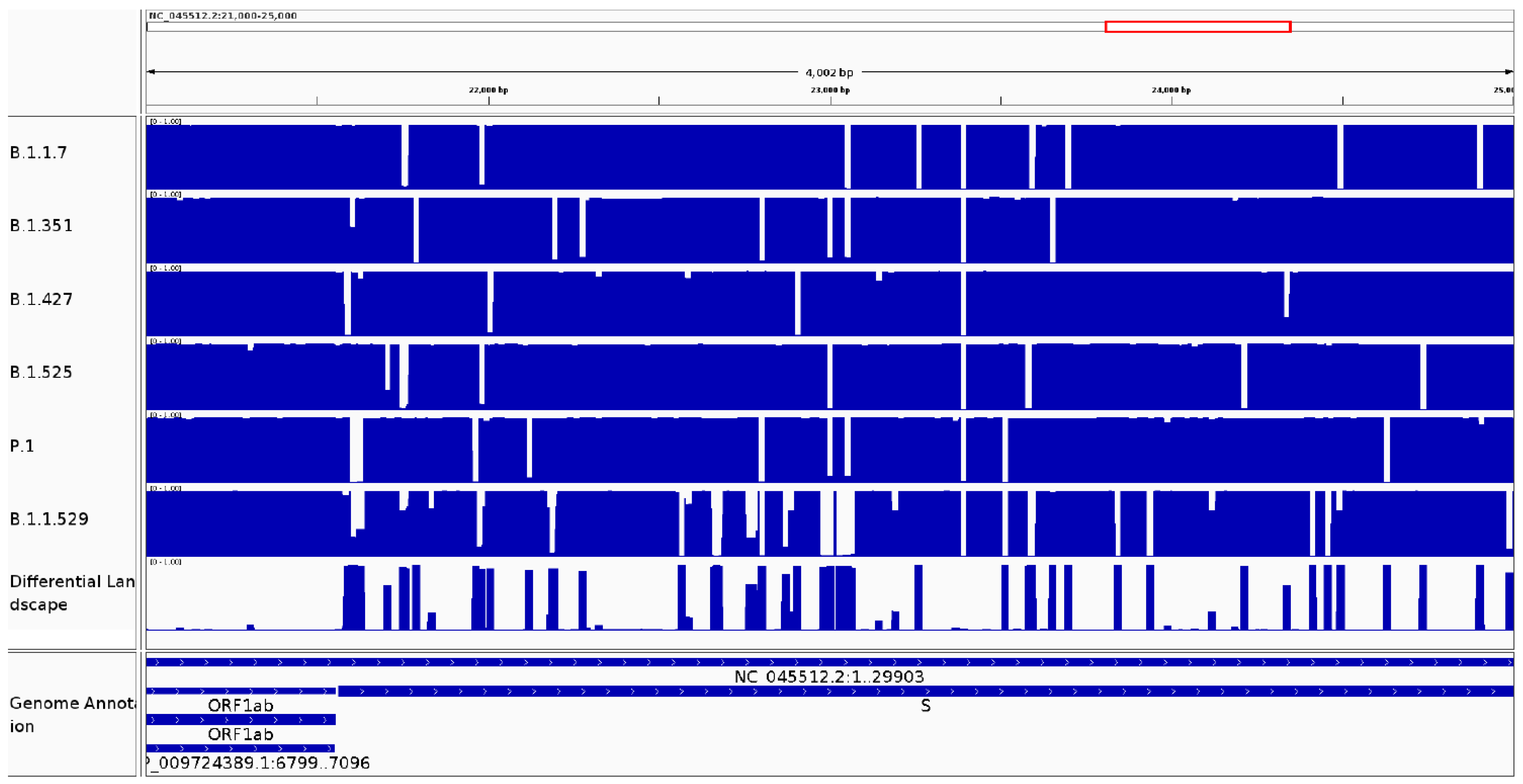
3.2.2. It Can Be Used to Identify Variable Regions between Phylogenetic Clades of the Same Species
3.2.3. It Can Be Used to Identify Variable Regions between Known Lineages of the Same Species
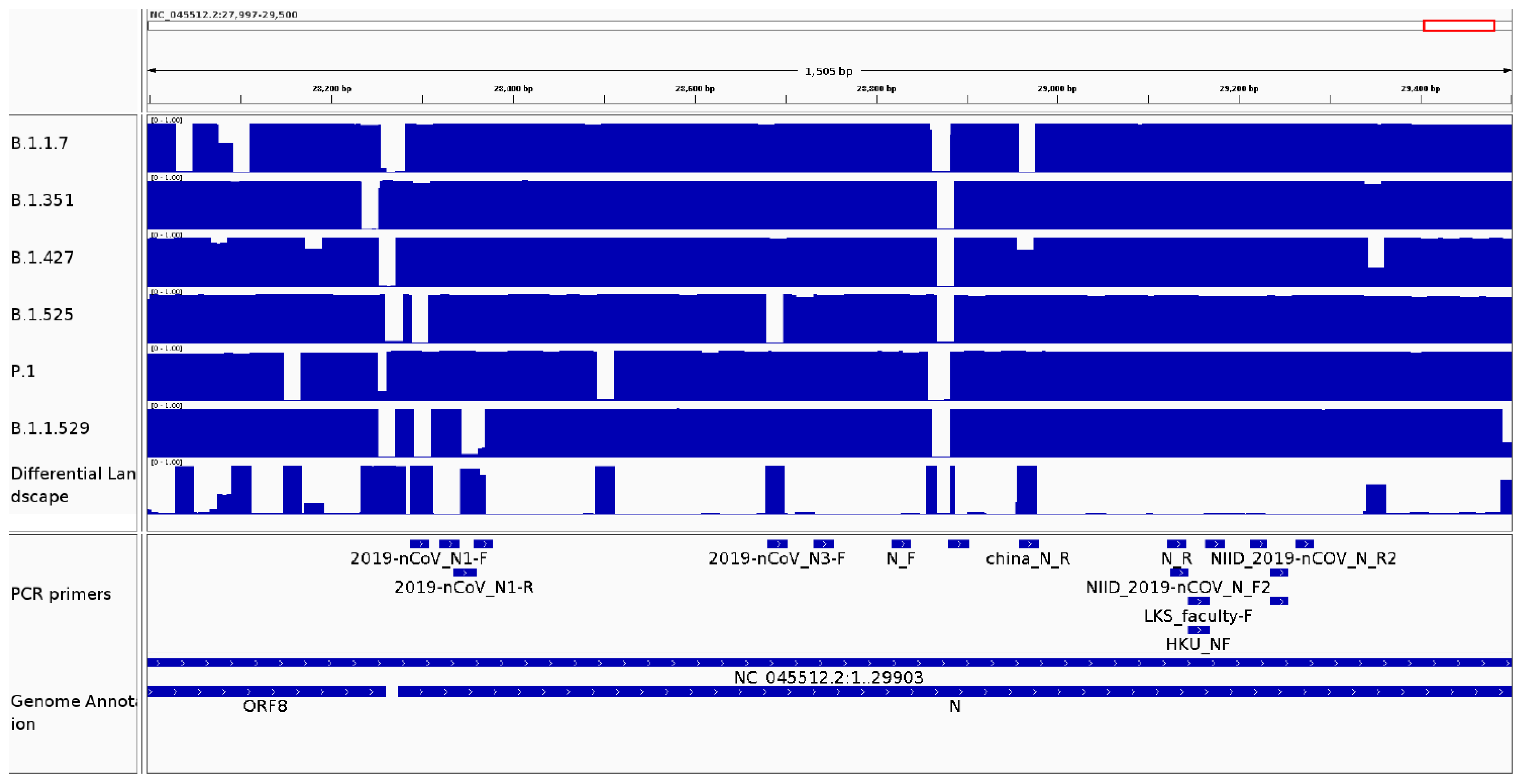
3.2.4. It Can Be Used as a Surveillance Tool to Monitor Genomic Variability in Primer and Probe Sequences from Diagnostic Kits
3.3. CovDif Can Generate a Conservation Landscape from a List of Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the Numbers. eLife 2020, 9, e57309. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamed, S.M.; Elkhatib, W.F.; Khairalla, A.S.; Noreddin, A.M. Global Dynamics of SARS-CoV-2 Clades and Their Relation to COVID-19 Epidemiology. Sci. Rep. 2021, 11, 8435. [Google Scholar] [CrossRef]
- Available online: 2021_01_11_Transmissibility_and_severity_of_501Y_V2_in_SA.Pdf (accessed on 2 March 2022).
- CMMID COVID-19 Working Group; Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased Mortality in Community-Tested Cases of SARS-CoV-2 Lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Guan, W.-D.; Chen, L.-P.; Ye, F.; Ye, D.; Wu, S.-G.; Zhou, H.-X.; He, J.-Y.; Yang, C.-G.; Zeng, Z.-Q.; Wang, Y.-T.; et al. High-Throughput Sequencing for Confirmation of Suspected 2019-NCoV Infection Identified by Fluorescence Quantitative Polymerase Chain Reaction. Chin. Med. J. 2020, 133, 1385–1386. [Google Scholar] [CrossRef]
- Li, D.; Zhang, J.; Li, J. Primer Design for Quantitative Real-Time PCR for the Emerging Coronavirus SARS-CoV-2. Theranostics 2020, 10, 7150–7162. [Google Scholar] [CrossRef]
- Park, M.; Won, J.; Choi, B.Y.; Lee, C.J. Optimization of Primer Sets and Detection Protocols for SARS-CoV-2 of Coronavirus Disease 2019 (COVID-19) Using PCR and Real-Time PCR. Exp. Mol. Med. 2020, 52, 963–977. [Google Scholar] [CrossRef]
- Venter, M.; Richter, K. Towards Effective Diagnostic Assays for COVID-19: A Review. J. Clin. Pathol. 2020, 73, 370–377. [Google Scholar] [CrossRef]
- Olsen, L.R.; Simon, C.; Kudahl, U.J.; Bagger, F.O.; Winther, O.; Reinherz, E.L.; Zhang, G.L.; Brusic, V. A Computational Method for Identification of Vaccine Targets from Protein Regions of Conserved Human Leukocyte Antigen Binding. BMC Med. Genom. 2015, 8, S1. [Google Scholar] [CrossRef] [Green Version]
- Pollard, A.J.; Bijker, E.M. A Guide to Vaccinology: From Basic Principles to New Developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-N.; Hsu, W.-L. GSAlign: An Efficient Sequence Alignment Tool for Intra-Species Genomes. BMC Genom. 2020, 21, 182. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Minkin, I.; Medvedev, P. Scalable Multiple Whole-Genome Alignment and Locally Collinear Block Construction with SibeliaZ. Nat. Commun. 2020, 11, 6327. [Google Scholar] [CrossRef]
- Moshiri, N. ViralMSA: Massively Scalable Reference-Guided Multiple Sequence Alignment of Viral Genomes. Bioinformatics 2021, 37, 714–716. [Google Scholar] [CrossRef]
- Mullen, J.L.; Tsueng, G.; Latif, A.A.; Alkuzweny, M.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. Hughes, and the Center for Viral Systems Biology. Available online: https://outbreak.info/2020 (accessed on 2 March 2022).
- Yip, C.C.-Y.; Ho, C.-C.; Chan, J.F.-W.; To, K.K.-W.; Chan, H.S.-Y.; Wong, S.C.-Y.; Leung, K.-H.; Fung, A.Y.-F.; Ng, A.C.-K.; Zou, Z.; et al. Development of a Novel, Genome Subtraction-Derived, SARS-CoV-2-Specific COVID-19-Nsp2 Real-Time RT-PCR Assay and Its Evaluation Using Clinical Specimens. Int. J. Mol. Sci. 2020, 21, 2574. [Google Scholar] [CrossRef] [Green Version]
- Dasaraju, P.V.; Liu, C. Infections of the Respiratory System. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Pabbaraju, K.; Wong, S.; Wong, A.A.; Appleyard, G.D.; Chui, L.; Pang, X.-L.; Yanow, S.K.; Fonseca, K.; Lee, B.E.; Fox, J.D.; et al. Design and Validation of Real-Time Reverse Transcription-PCR Assays for Detection of Pandemic (H1N1) 2009 Virus. J. Clin. Microbiol. 2009, 47, 3454–3460. [Google Scholar] [CrossRef] [Green Version]
- Quest Diagnostics Respiratory Viral Panel, PCR. 2021. Available online: https://testdirectory.questdiagnostics.com/test/test-detail/95512/respiratory-viral-panel-pcr?cc=MASTER (accessed on 2 March 2022).
- Weese, D.; Holtgrewe, M.; Reinert, K. RazerS 3: Faster, Fully Sensitive Read Mapping. Bioinformatics 2012, 28, 2592–2599. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Tange, O. GNU Parallel 2018; Ole Tange: Hørsholm, Denmark, 2018. [Google Scholar]
- GISAID Clade and Lineage Nomenclature. 2 March 2021. Available online: https://www.gisaid.org/resources/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/ (accessed on 2 March 2022).
- PANGO Lineages: Latest Epidemiological Lineages of SARS-CoV-2021. Available online: https://cov-lineages.org/ (accessed on 2 March 2022).
- World Health Organization. Detection of 2019 Novel Coronavirus (2019-NCoV) in Suspected Human Cases by RT-PCR 2021. Available online: https://www.who.int/docs/default-source/coronaviruse/peiris-protocol-16-1-20.pdf (accessed on 2 March 2022).
- Centers for Disease Control and Prevention. Research Use Only 2019-Novel Coronavirus (2019-NCoV) Real-Time RT-PCR Primers and Probes. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed on 2 March 2022).
- Bio-Basic COVID-19 Primers/Probes Extraction Detection Kits. Available online: https://www.biobasic.com/coronavirus-covid-19-primers/ (accessed on 2 March 2022).
- Mollaei, H.R.; Afshar, A.A.; Kalantar-Neyestanaki, D.; Fazlalipour, M.; Aflatoonian, B. Comparison Five Primer Sets from Different Genome Region of COVID-19 for Detection of Virus Infection by Conventional RT-PCR. Iran. J. Microbiol. 2020, 12, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Park, G.-S.; Moon, J.H.; Ku, K.; Beak, S.-H.; Lee, C.-S.; Kim, S.; Park, E.C.; Park, D.; Lee, J.-H.; et al. Comparative Analysis of Primer–Probe Sets for RT-QPCR of COVID-19 Causative Virus (SARS-CoV-2). ACS Infect. Dis. 2020, 6, 2513–2523. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Arena, F.; Pollini, S.; Rossolini, G.M.; Margaglione, M. Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic. Int. J. Mol. Sci. 2021, 22, 1298. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No Evidence for Increased Transmissibility from Recurrent Mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Saha, I.; Ghosh, N.; Maity, D.; Sharma, N.; Sarkar, J.P.; Mitra, K. Genome-Wide Analysis of Indian SARS-CoV-2 Genomes for the Identification of Genetic Mutation and SNP. Infect. Genet. Evol. 2020, 85, 104457. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, G.; Meng, K.; Yang, L.; Liu, D.; Meng, G. Rapid Spread of Mutant Alleles in Worldwide SARS-CoV-2 Strains Revealed by Genome-Wide Single Nucleotide Polymorphism and Variation Analysis. Genome Biol. Evol. 2021, 13, evab015. [Google Scholar] [CrossRef]
- Kumar, R.; Verma, H.; Singhvi, N.; Sood, U.; Gupta, V.; Singh, M.; Kumari, R.; Hira, P.; Nagar, S.; Talwar, C.; et al. Comparative Genomic Analysis of Rapidly Evolving SARS-CoV-2 Reveals Mosaic Pattern of Phylogeographical Distribution. mSystems 2020, 5, e00505-20. [Google Scholar] [CrossRef]
- Yin, C. Genotyping Coronavirus SARS-CoV-2: Methods and Implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--a Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Rangan, R.; Zheludev, I.N.; Das, R. RNA Genome Conservation and Secondary Structure in SARS-CoV-2 and SARS-Related Viruses. RNA 2020, 26, 937–959. [Google Scholar] [CrossRef]
- Anantharajah, A.; Helaers, R.; Defour, J.-P.; Olive, N.; Kabera, F.; Croonen, L.; Deldime, F.; Vaerman, J.-L.; Barbée, C.; Bodéus, M.; et al. How to Choose the Right Real-Time RT-PCR Primer Sets for the SARS-CoV-2 Genome Detection? J. Virol. Methods 2021, 295, 114197. [Google Scholar] [CrossRef] [PubMed]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 Mutation Hot Spots Include a Novel RNA-Dependent-RNA Polymerase Variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging SARS-CoV-2 Variants and Impact in Global Vaccination Programs against SARS-CoV-2/COVID-19. Vaccines 2021, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Liu, S.; Shen, J.; Lu, A.Z.; Zhang, Y.; Li, K.; Liu, J.; Yang, L.; Hu, C.-D.; Wan, J. Updated SARS-CoV-2 Single Nucleotide Variants and Mortality Association. J. Med. Virol. 2021, 93, 6525–6534. [Google Scholar] [CrossRef]
- Klungthong, C.; Chinnawirotpisan, P.; Hussem, K.; Phonpakobsin, T.; Manasatienkij, W.; Ajariyakhajorn, C.; Rungrojcharoenkit, K.; Gibbons, R.V.; Jarman, R.G. The Impact of Primer and Probe-Template Mismatches on the Sensitivity of Pandemic Influenza A/H1N1/2009 Virus Detection by Real-Time RT-PCR. J. Clin. Virol. 2010, 48, 91–95. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.M.; Schutten, M. The Effect of Primer-Template Mismatches on the Detection and Quantification of Nucleic Acids Using the 5′ Nuclease Assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cedeño-Pérez, L.F.; Gómez-Romero, L. CovDif, a Tool to Visualize the Conservation between SARS-CoV-2 Genomes and Variants. Viruses 2022, 14, 561. https://doi.org/10.3390/v14030561
Cedeño-Pérez LF, Gómez-Romero L. CovDif, a Tool to Visualize the Conservation between SARS-CoV-2 Genomes and Variants. Viruses. 2022; 14(3):561. https://doi.org/10.3390/v14030561
Chicago/Turabian StyleCedeño-Pérez, Luis F., and Laura Gómez-Romero. 2022. "CovDif, a Tool to Visualize the Conservation between SARS-CoV-2 Genomes and Variants" Viruses 14, no. 3: 561. https://doi.org/10.3390/v14030561
APA StyleCedeño-Pérez, L. F., & Gómez-Romero, L. (2022). CovDif, a Tool to Visualize the Conservation between SARS-CoV-2 Genomes and Variants. Viruses, 14(3), 561. https://doi.org/10.3390/v14030561