
Conserved Targets to Prevent Emerging Coronaviruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
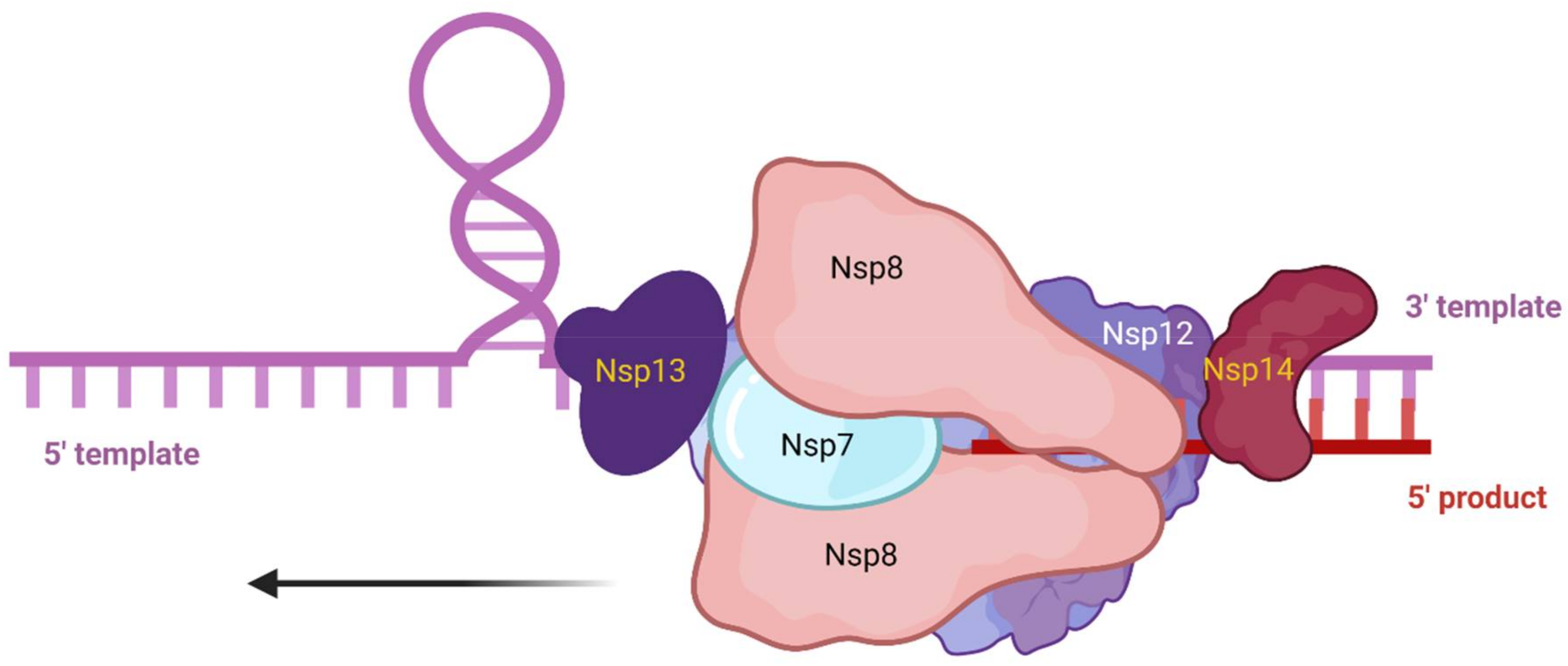{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
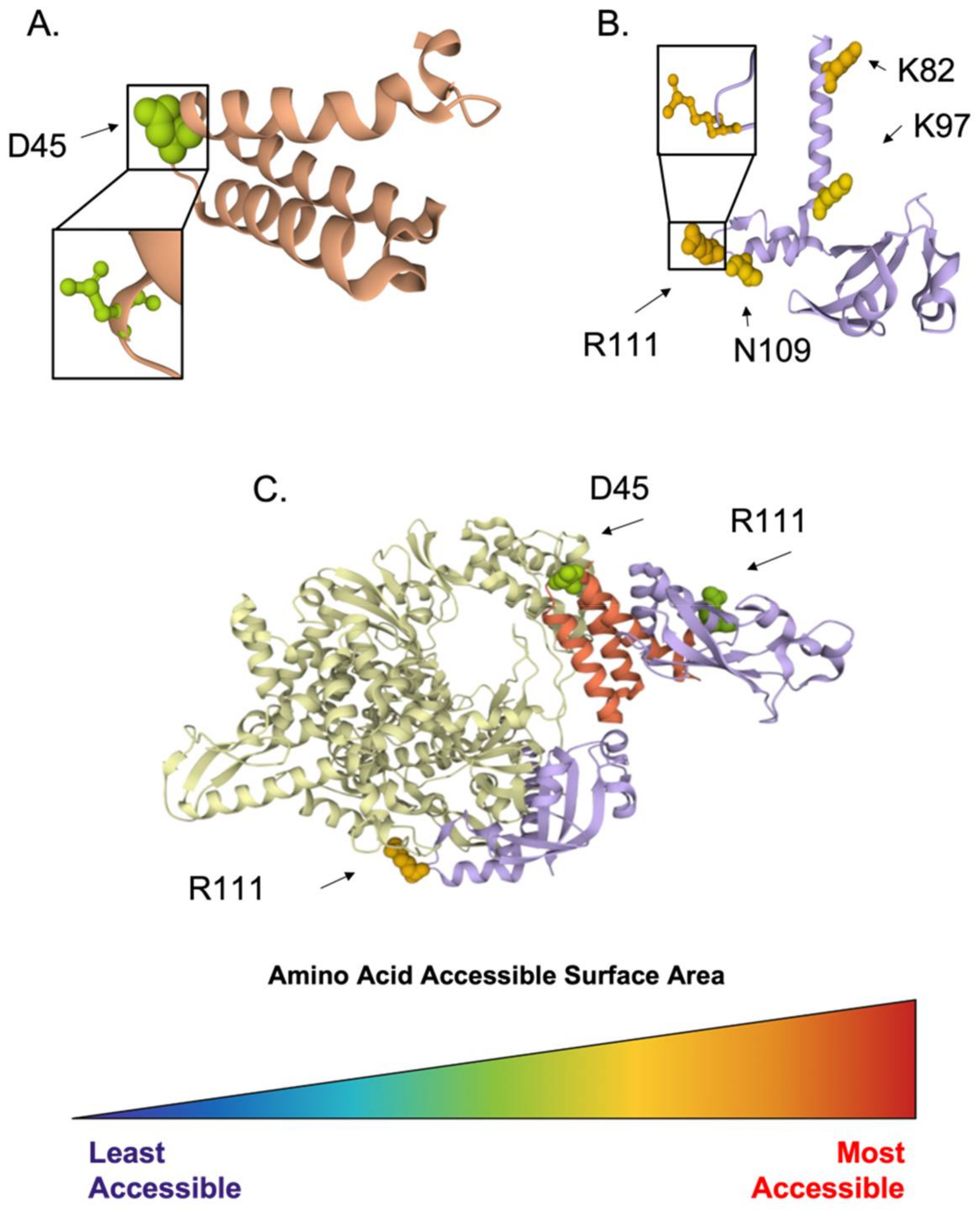
3.1. Conserved Antiviral Targets in Coronavirus Molecular Life Cycle
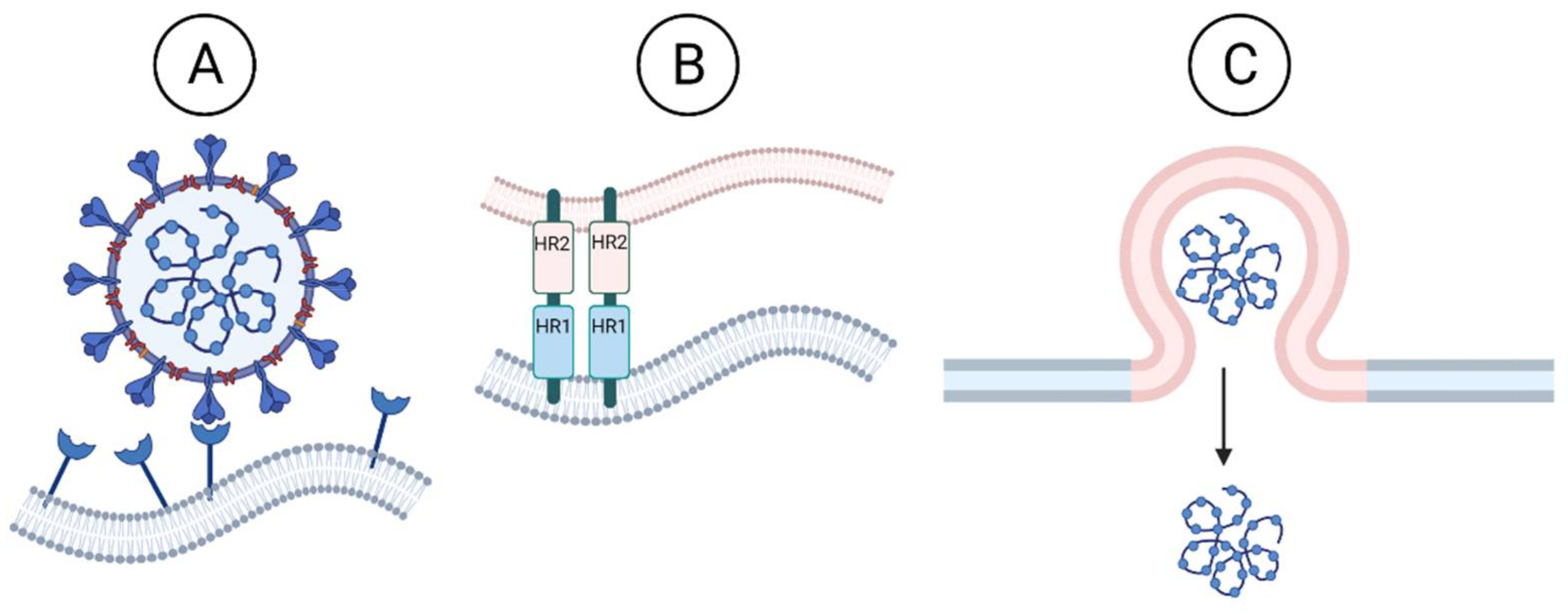
3.1.1. Inhibition of Viral Entry
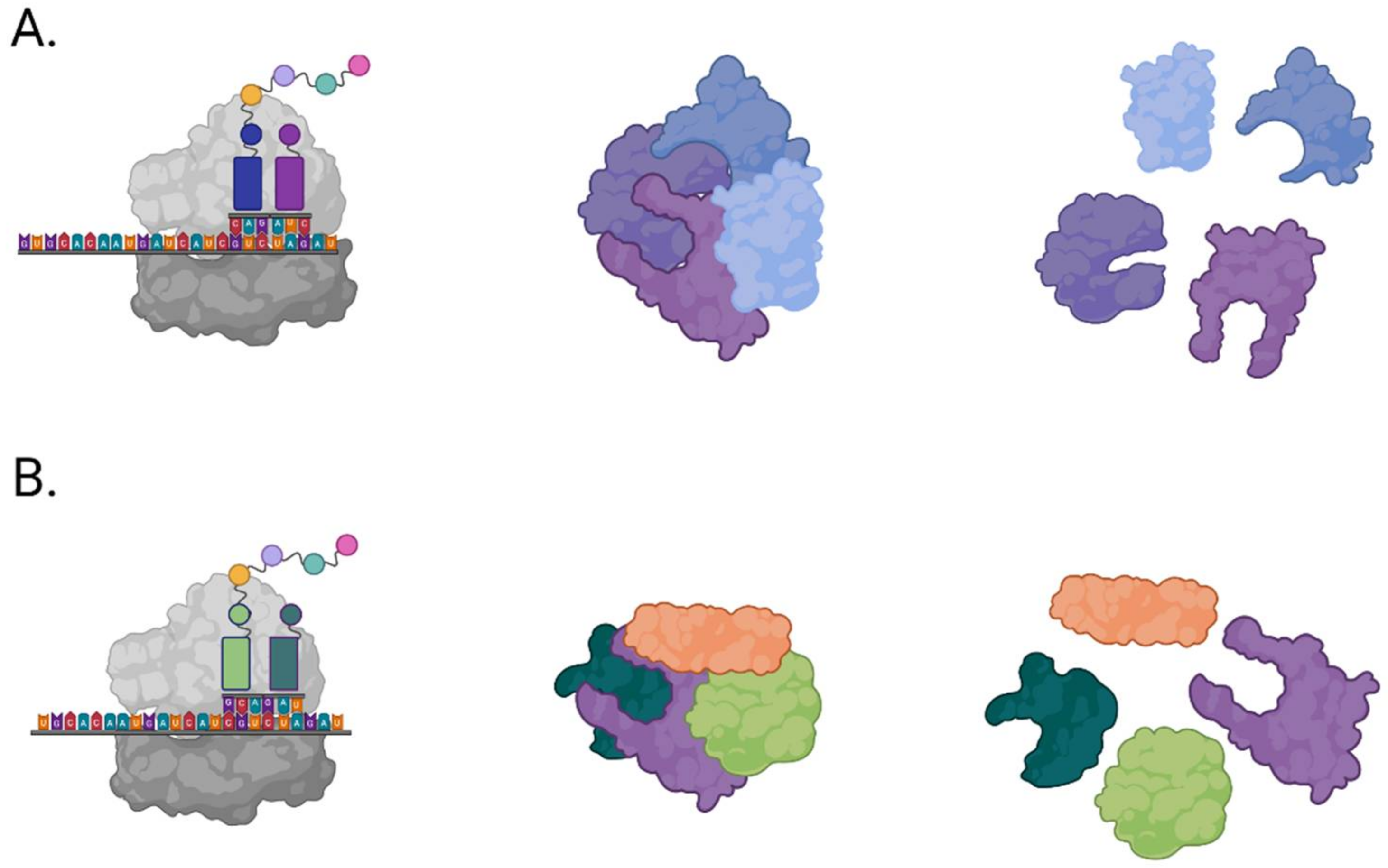
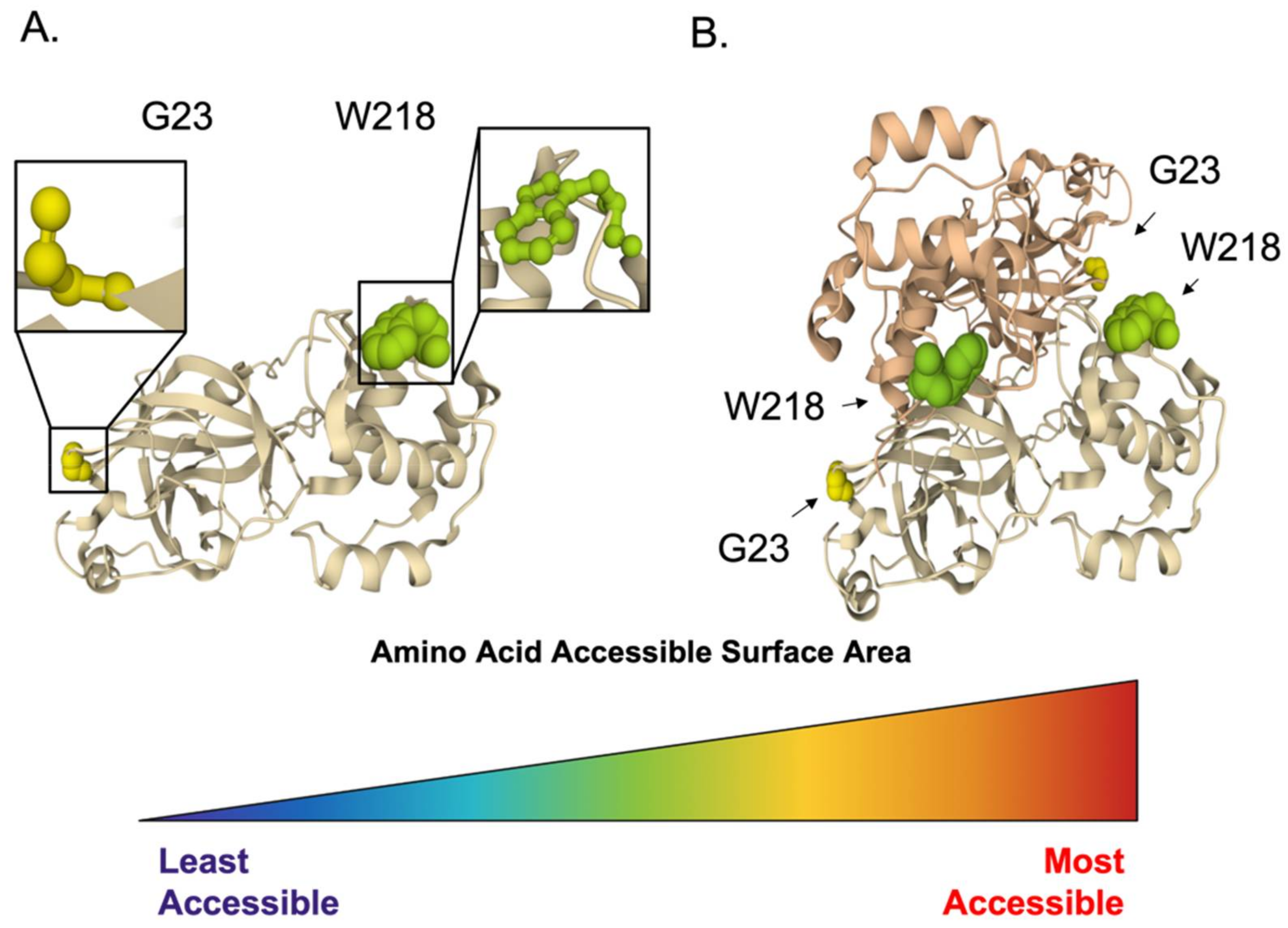
3.1.2. Inhibition of Viral Proteases
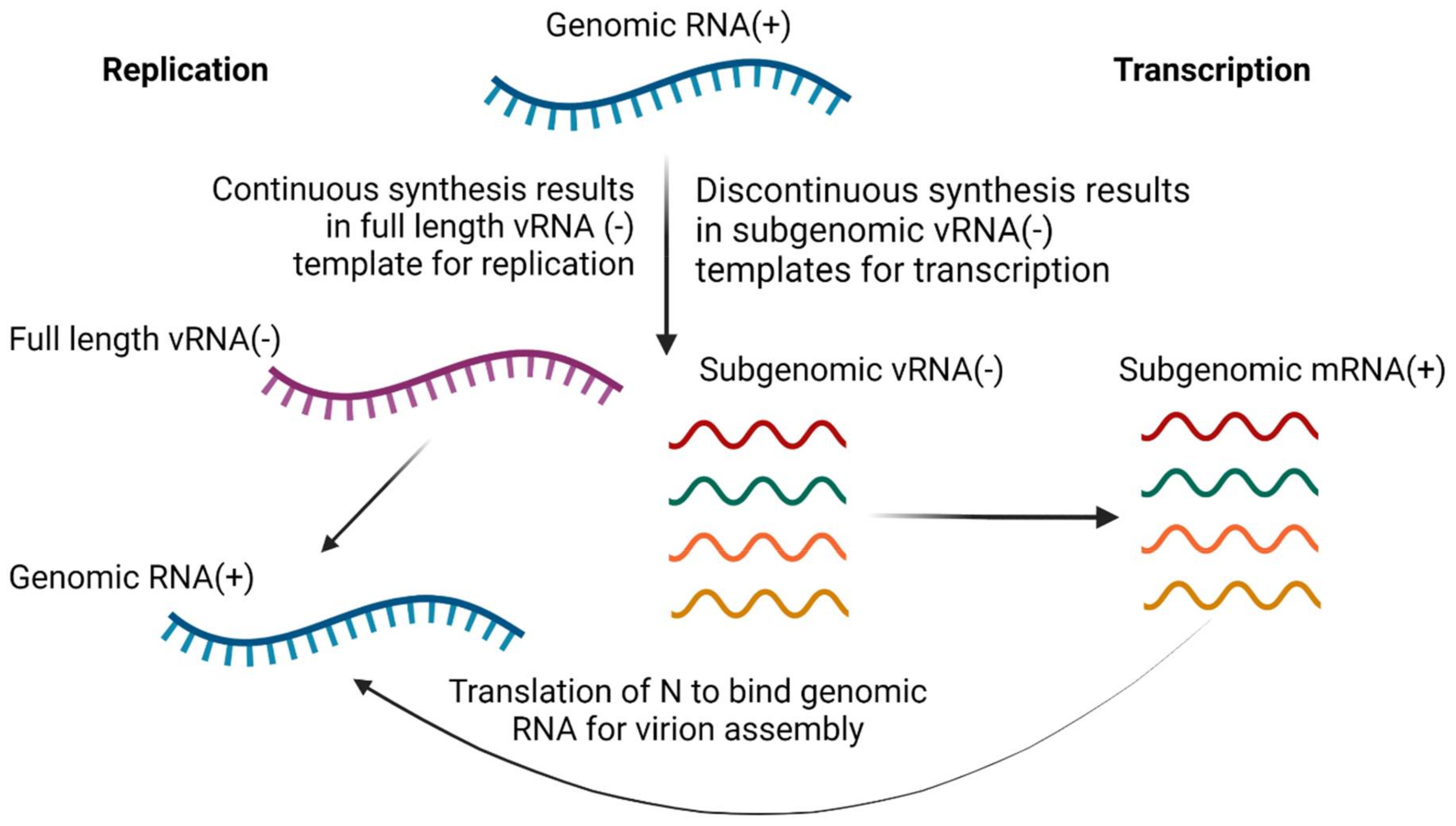
3.1.3. Inhibition of Transcription and Replication
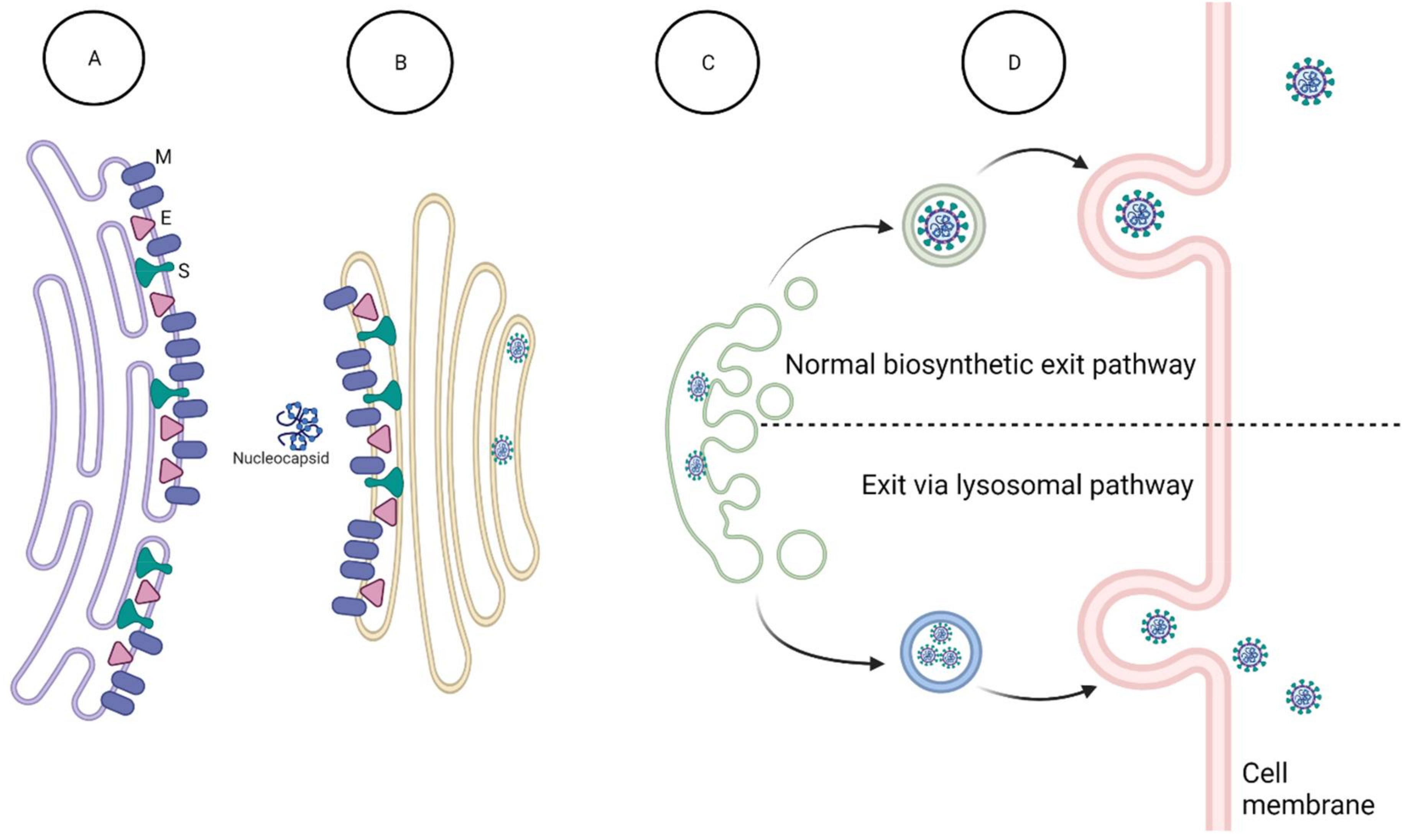
3.1.4. Inhibition of Virion Assembly and Exit
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- COVID-19 Data Explorer. Available online: https://ourworldindata.org/coronavirus-data-explorer (accessed on 3 March 2022).
- Shaman, J.; Galanti, M. Will SARS-CoV-2 become endemic? Science 2020, 370, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Past Seasons Vaccine Effectiveness Estimates|CDC. Available online: https://www.cdc.gov/flu/vaccines-work/past-seasons-estimates.html (accessed on 8 February 2022).
- El Sahly, H.M.; Baden, L.R.; Essink, B.; Doblecki-Lewis, S.; Martin, J.M.; Anderson, E.J.; Campbell, T.B.; Clark, J.; Jackson, L.A.; Fichtenbaum, C.J.; et al. Efficacy of the mRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N. Engl. J. Med. 2021, 385, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; St. Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- Thompson, M.G. Effectiveness of a Third Dose of mRNA Vaccines Against COVID-19–Associated Emergency Department and Urgent Care Encounters and Hospitalizations Among Adults During Periods of Delta and Omicron Variant Predominance—VISION Network, 10 States, August 2021–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 139–145. [Google Scholar] [CrossRef]
- Wu, M.; Wall, E.C.; Carr, E.J.; Harvey, R.; Townsley, H.; Mears, H.V.; Adams, L.; Kjaer, S.; Kelly, G.; Warchal, S.; et al. Three-dose vaccination elicits neutralising antibodies against omicron. Lancet 2022, 399, 715–717. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Ten Health Issues WHO Will Tackle This Year. Available online: https://www.who.int/news-room/spotlight/ten-threats-to-global-health-in-2019 (accessed on 26 August 2021).
- Virus Pathogen Database and Analysis Resource (ViPR)-Genome Database with Visualization and Analysis Tools. Available online: https://www.viprbrc.org/brc/home.spg?decorator=vipr (accessed on 26 August 2021).
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Protein Data Bank. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 26 August 2021).
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Ahmad, S.; Gromiha, M.; Fawareh, H.; Sarai, A. ASAView: Database and tool for solvent accessibility representation in proteins. BMC Bioinform. 2004, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The MERS-CoV Receptor DPP4 as a Candidate Binding Target of the SARS-CoV-2 Spike. iScience 2020, 23, 101160. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Mudgal, G.; Santiago, C.; Casasnovas, J.M. A structural view of coronavirus–receptor interactions. Virus Res. 2014, 194, 3–15. [Google Scholar] [CrossRef]
- Wang, G.; Yang, M.-L.; Duan, Z.-L.; Liu, F.-L.; Jin, L.; Long, C.-B.; Zhang, M.; Tang, X.-P.; Xu, L.; Li, Y.-C.; et al. Dalbavancin binds ACE2 to block its interaction with SARS-CoV-2 spike protein and is effective in inhibiting SARS-CoV-2 infection in animal models. Cell Res. 2021, 31, 17–24. [Google Scholar] [CrossRef]
- REGEN-COVTM Antibody Cocktail Is Active Against SARS-CoV-2 Variants First Identified in the UK and South Africa|Regeneron Pharmaceuticals Inc. Available online: https://investor.regeneron.com/index.php/news-releases/news-release-details/regen-covtm-antibody-cocktail-active-against-sars-cov-2-variants/ (accessed on 26 August 2021).
- Bamlanivimab and Etesevimab EUA|Lilly COVID-19 Products. Available online: https://www.covid19.lilly.com/bam-ete (accessed on 8 February 2022).
- CoVariants. Available online: https://covariants.org/variants/21K.Omicron (accessed on 9 February 2022).
- Cavazzoni, P. (COVID-19) Update: FDA Limits Use of Certain Monoclonal Antibodies to Treat COVID-19 Due to the Omicron Variant. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-certain-monoclonal-antibodies-treat-covid-19-due-omicron (accessed on 8 February 2022).
- Wang, X.; Xia, S.; Zhu, Y.; Lu, L.; Jiang, S. Pan-coronavirus fusion inhibitors as the hope for today and tomorrow. Protein Cell 2021, 12, 84–88. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Eggink, D.; Bontjer, I.; de Taeye, S.W.; Langedijk, J.P.M.; Berkhout, B.; Sanders, R.W. HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J. Biol. Chem. 2019, 294, 5736–5746. [Google Scholar] [CrossRef]
- Lim, K.P.; Ng, L.F.P.; Liu, D.X. Identification of a Novel Cleavage Activity of the First Papain-Like Proteinase Domain Encoded by Open Reading Frame 1a of the Coronavirus Avian Infectious Bronchitis Virus and Characterization of the Cleavage Products. J. Virol. 2000, 74, 1674–1685. [Google Scholar] [CrossRef]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef]
- Mahdi, M. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2’s main protease. J. Virol. 2020, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 2020, 12, eabc5332. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Seeks Emergency Use Authorization for Novel COVID-19 Oral Antiviral Candidate|Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-seeks-emergency-use-authorization-novel-covid-19 (accessed on 5 December 2021).
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Dwivedy, A.; Mariadasse, R.; Ahmad, M.; Chakraborty, S.; Kar, D.; Tiwari, S.; Majumdar, T.; Jeyakanthan, J.; Biswal, B.K. Characterization of the NiRAN domain from RNA-dependent RNA polymerase provides insights into a potential therapeutic target against SARS-CoV-2. PLoS Comput. Biol. 2021, 17, e1009384. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef]
- Ivanov, K.A.; Ziebuhr, J. Human Coronavirus 229E Nonstructural Protein 13: Characterization of Duplex-Unwinding, Nucleoside Triphosphatase, and RNA 5′-Triphosphatase Activities. J. Virol. 2004, 78, 6. [Google Scholar] [CrossRef]
- Tvarogová, J.; Madhugiri, R.; Bylapudi, G.; Ferguson, L.J.; Karl, N.; Ziebuhr, J. Identification and Characterization of a Human Coronavirus 229E Nonstructural Protein 8-Associated RNA 3′-Terminal Adenylyltransferase Activity. J. Virol. 2019, 93, e00291-19. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio. 2018, 9, e00221-18. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; De Jonghe, S.; Maes, P.; Weynand, B.; Neyts, J. Molnupiravir Inhibits Replication of the Emerging SARS-CoV-2 Variants of Concern in a Hamster Infection Model. J. Infect. Dis. 2021, 224, 749–753. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Solanki, K.; Li, Y.; Ma, Z.; Peppelenbosch, M.P.; Baig, M.S.; Pan, Q. Viral polymerase binding and broad-spectrum antiviral activity of molnupiravir against human seasonal coronaviruses. Virology 2021, 564, 33–38. [Google Scholar] [CrossRef]
- Merck and Ridgeback’s Investigational Oral Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50 Percent Compared to Placebo for Patients with Mild or Moderate COVID-19 in Positive Interim Analysis of Phase 3 Study. Available online: https://www.merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalization-or-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat/ (accessed on 13 October 2021).
- Merck and Ridgeback Announce Submission of Emergency Use Authorization Application to the U.S. FDA for Molnupiravir, an Investigational Oral Antiviral Medicine, for the Treatment of Mild-to-Moderate COVID-19 in At Risk Adults. Available online: https://www.merck.com/news/merck-and-ridgeback-announce-submission-of-emergency-use-authorization-application-to-the-u-s-fda-for-molnupiravir-an-investigational-oral-antiviral-medicine-for-the-treatment-of-mild-to-moderate-c/ (accessed on 13 October 2021).
- Cavazzoni, P. Coronavirus (COVID-19) Update: FDA Authorizes Additional Oral Antiviral for Treatment of COVID-19 in Certain Adults. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain (accessed on 8 February 2022).
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Spratt, A.N.; Gallazzi, F.; Quinn, T.P.; Lorson, C.L.; Sönnerborg, A.; Singh, K. Coronavirus helicases: Attractive and unique targets of antiviral drug-development and therapeutic patents. Expert Opin. Ther. Pat. 2021, 31, 339–350. [Google Scholar] [CrossRef]
- Shu, T.; Huang, M.; Wu, D.; Ren, Y.; Zhang, X.; Han, Y.; Mu, J.; Wang, R.; Qiu, Y.; Zhang, D.-Y.; et al. SARS-Coronavirus-2 Nsp13 Possesses NTPase and RNA Helicase Activities That Can Be Inhibited by Bismuth Salts. Virol. Sin. 2020, 35, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Seybert, A.; Posthuma, C.C.; van Dinten, L.C.; Snijder, E.J.; Gorbalenya, A.E.; Ziebuhr, J. A Complex Zinc Finger Controls the Enzymatic Activities of Nidovirus Helicases. J. Virol. 2005, 79, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Yosaatmadja, Y.; Douangamath, A.; Arrowsmith, C.H.; von Delft, F.; Edwards, A.; Bountra, C.; Gileadi, O. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021, 12, 4848. [Google Scholar] [CrossRef]
- Wu, C.-H.; Chen, P.-J.; Yeh, S.-H. Nucleocapsid Phosphorylation and RNA Helicase DDX1 Recruitment Enables Coronavirus Transition from Discontinuous to Continuous Transcription. Cell Host Microbe 2014, 16, 462–472. [Google Scholar] [CrossRef]
- Chang, C.; Michalska, K.; Jedrzejczak, R.; Maltseva, N.; Endres, M.; Godzik, A.; Kim, Y.; Joachimiak, A. Crystal structure of RNA-binding domain of nucleocapsid phosphoprotein from SARS-CoV-2, monoclinic crystal form. Wordwide PDB 2021. [Google Scholar] [CrossRef]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef] [PubMed]
- McBride, C.E.; Li, J.; Machamer, C.E. The Cytoplasmic Tail of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Contains a Novel Endoplasmic Reticulum Retrieval Signal That Binds COPI and Promotes Interaction with Membrane Protein. J. Virol. 2007, 81, 2418–2428. [Google Scholar] [CrossRef]
- Oostra, M.; de Haan, C.A.M.; de Groot, R.J.; Rottier, P.J.M. Glycosylation of the Severe Acute Respiratory Syndrome Coronavirus Triple-Spanning Membrane Proteins 3a and M. J. Virol. 2006, 80, 2326–2336. [Google Scholar] [CrossRef]
- Corse, E.; Machamer, C.E. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 2003, 312, 25–34. [Google Scholar] [CrossRef]
- Narayanan, K.; Maeda, A.; Maeda, J.; Makino, S. Characterization of the Coronavirus M Protein and Nucleocapsid Interaction in Infected Cells. J. Virol. 2000, 74, 8127–8134. [Google Scholar] [CrossRef]
- Zinzula, L.; Basquin, J.; Bohn, S.; Beck, F.; Klumpe, S.; Pfeifer, G.; Nagy, I.; Bracher, A.; Hartl, F.U.; Baumeister, W. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the COVID-19 severe acute respiratory syndrome coronavirus 2. Biochem. Biophys. Res. Commun. 2021, 538, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kim, J.; Park, S.; Kim, D.; Kim, M.; Baek, K.; Bae, J.-Y.; Park, M.-S.; Kim, W.-K.; Lee, Y.; et al. MERS-CoV and SARS-CoV-2 replication can be inhibited by targeting the interaction between the viral spike protein and the nucleocapsid protein. Theranostics 2021, 11, 3853–3867. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Protein Structure Prediction by Trrosetta. Available online: https://yanglab.nankai.edu.cn/trRosetta/ (accessed on 13 October 2021).
- Mahtarin, R.; Islam, S.; Islam, J.; Ullah, M.O.; Ali, M.A.; Halim, M.A. Structure and dynamics of membrane protein in SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–14, ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Jiang, S.; Yang, C.; Liu, N.; Dai, H.; Lee, I.; Meng, X.; Yuan, Z. Probing the formation, structure and free energy relationships of M protein dimers of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef]
- Tooze, J.; Tooze, S.A. Sorting of Progeny Coronavirus from Condensed Secretory Proteins at the Exit from the Trans-GolNetwork of AtT20 Cells. J. Cell Biol. 1987, 105, 1215–1226. [Google Scholar] [CrossRef]
- Ravindran, M.S.; Bagchi, P.; Cunningham, C.N.; Tsai, B. Opportunistic intruders: How viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol. 2016, 14, 407–420. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef]
- Gorshkov, K.; Chen, C.Z.; Bostwick, R.; Rasmussen, L.; Tran, B.N.; Cheng, Y.-S.; Xu, M.; Pradhan, M.; Henderson, M.; Zhu, W.; et al. The SARS-CoV-2 Cytopathic Effect Is Blocked by Lysosome Alkalizing Small Molecules. ACS Infect. Dis. 2021, 7, 1389–1408. [Google Scholar] [CrossRef] [PubMed]
- Eguia, R.T.; Crawford, K.H.D.; Stevens-Ayers, T.; Kelnhofer-Millevolte, L.; Greninger, A.L.; Englund, J.A.; Boeckh, M.J.; Bloom, J.D. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021, 17, e1009453. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Eaton, B.T. Bats, Civets and the Emergence of SARS. In Wildlife and Emerging Zoonotic Diseases: The Biology, Circumstances and Consequences of Cross-Species Transmission; Childs, J.E., Mackenzie, J.S., Richt, J.A., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2007; Volume 315, pp. 325–344. ISBN 978-3-540-70961-9. [Google Scholar]
- Omrani, A.S.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East respiratory syndrome coronavirus (MERS-CoV): Animal to human interaction. Pathog. Glob. Health 2015, 109, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- van der Vries, E.; Veldhuis Kroeze, E.J.; Stittelaar, K.J.; Linster, M.; Van der Linden, A.; Schrauwen, E.J.A.; Leijten, L.M.; van Amerongen, G.; Schutten, M.; Kuiken, T.; et al. Multidrug Resistant 2009 A/H1N1 Influenza Clinical Isolate with a Neuraminidase I223R Mutation Retains Its Virulence and Transmissibility in Ferrets. PLoS Pathog. 2011, 7, e1002276. [Google Scholar] [CrossRef]
- Davis, A.M.; Chabolla, B.J.; Newcomb, L.L. Emerging antiviral resistant strains of influenza A and the potential therapeutic targets within the viral ribonucleoprotein (vRNP) complex. Virol. J. 2014, 11, 167. [Google Scholar] [CrossRef][Green Version]
- Birnkrant, D. FDA Approves New Drug to Treat Influenza. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treat-influenza (accessed on 26 August 2021).
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Beale, K.K.; Robinson, W.E. Combinations of reverse transcriptase, protease, and integrase inhibitors can be synergistic in vitro against drug-sensitive and RT inhibitor-resistant molecular clones of HIV-1. Antiviral Res. 2000, 46, 223–232. [Google Scholar] [CrossRef]
- De Pace, V.; Morelli, M.C.; Ravaioli, M.; Maggi, F.; Galli, S.; Vero, V.; Re, M.C.; Cescon, M.; Pistello, M. Efficacy, Safety, and Predictors of Direct-acting antivirals in Hepatitis C Virus Patients with Heterogeneous Liver Diseases. New Microbiol. 2019, 42, 189–196. [Google Scholar]
- National Progress Report 2025 Goal: Reduce HCV Deaths|CDC. Available online: https://www.cdc.gov/hepatitis/policy/npr/2021/NationalProgressReport-HepC-ReduceDeaths.htm (accessed on 3 March 2022).
- HIV-Related Death Rate in U.S. Fell by Half From 2010 to 2017 Press Release|CDC. Available online: https://www.cdc.gov/nchhstp/newsroom/2020/hiv-related-death-rate-press-release.html (accessed on 3 March 2022).
- Kozlov, M. Why scientists are racing to develop more COVID antivirals. Nature 2022, 601, 496. [Google Scholar] [CrossRef]
- Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S1097276520305189?token=5D3CA6E332C07F4D29B88229D508E67CE7B2B558C09CF935FAFA8E6709027A162583B4FD21FFAA71F55EE9BC4058629F&originRegion=us-east-1&originCreation=20220209194027 (accessed on 9 February 2022).
- Terrier, O.; Dilly, S.; Pizzorno, A.; Chalupska, D.; Humpolickova, J.; Bouřa, E.; Berenbaum, F.; Quideau, S.; Lina, B.; Fève, B.; et al. Antiviral Properties of the NSAID Drug Naproxen Targeting the Nucleoprotein of SARS-CoV-2 Coronavirus. Molecules 2021, 26, 2593. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez Lomeli, F.; Elmaraghy, N.; Castro, A.; Osuna Guerrero, C.V.; Newcomb, L.L. Conserved Targets to Prevent Emerging Coronaviruses. Viruses 2022, 14, 563. https://doi.org/10.3390/v14030563
Gonzalez Lomeli F, Elmaraghy N, Castro A, Osuna Guerrero CV, Newcomb LL. Conserved Targets to Prevent Emerging Coronaviruses. Viruses. 2022; 14(3):563. https://doi.org/10.3390/v14030563
Chicago/Turabian StyleGonzalez Lomeli, Fernanda, Nicole Elmaraghy, Anthony Castro, Claudia V. Osuna Guerrero, and Laura L. Newcomb. 2022. "Conserved Targets to Prevent Emerging Coronaviruses" Viruses 14, no. 3: 563. https://doi.org/10.3390/v14030563
APA StyleGonzalez Lomeli, F., Elmaraghy, N., Castro, A., Osuna Guerrero, C. V., & Newcomb, L. L. (2022). Conserved Targets to Prevent Emerging Coronaviruses. Viruses, 14(3), 563. https://doi.org/10.3390/v14030563