
Complete Genome Sequence of Herpes Simplex Virus 2 Strain G

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. HSV-2 Strain G Virus and Whole Viral Genome DNA Sequencing
2.2. Whole Viral Genome Assembly
2.3. Determination of Genome Termini of HSV-2 Strain G
2.4. Identification of Sequencing Reads from Four HSV-2 Genome Isomers
2.5. Analysis of ‘α’ Sequences
2.6. Determination of ‘α‘ Sequence Copy Number in HSV2 Strain G Genome
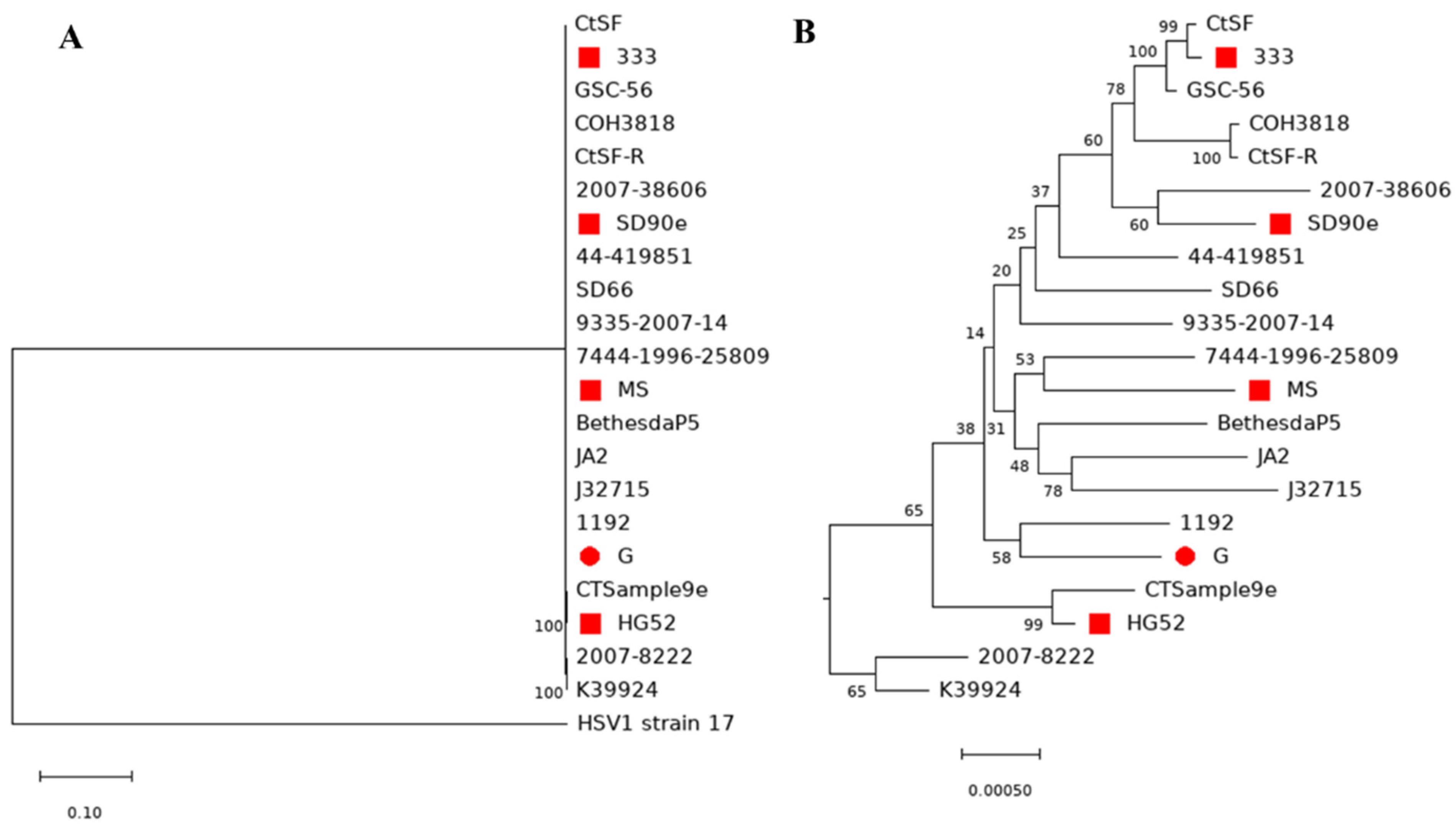
2.7. Sequence Comparison and Phylogenetic Analysis
3. Results
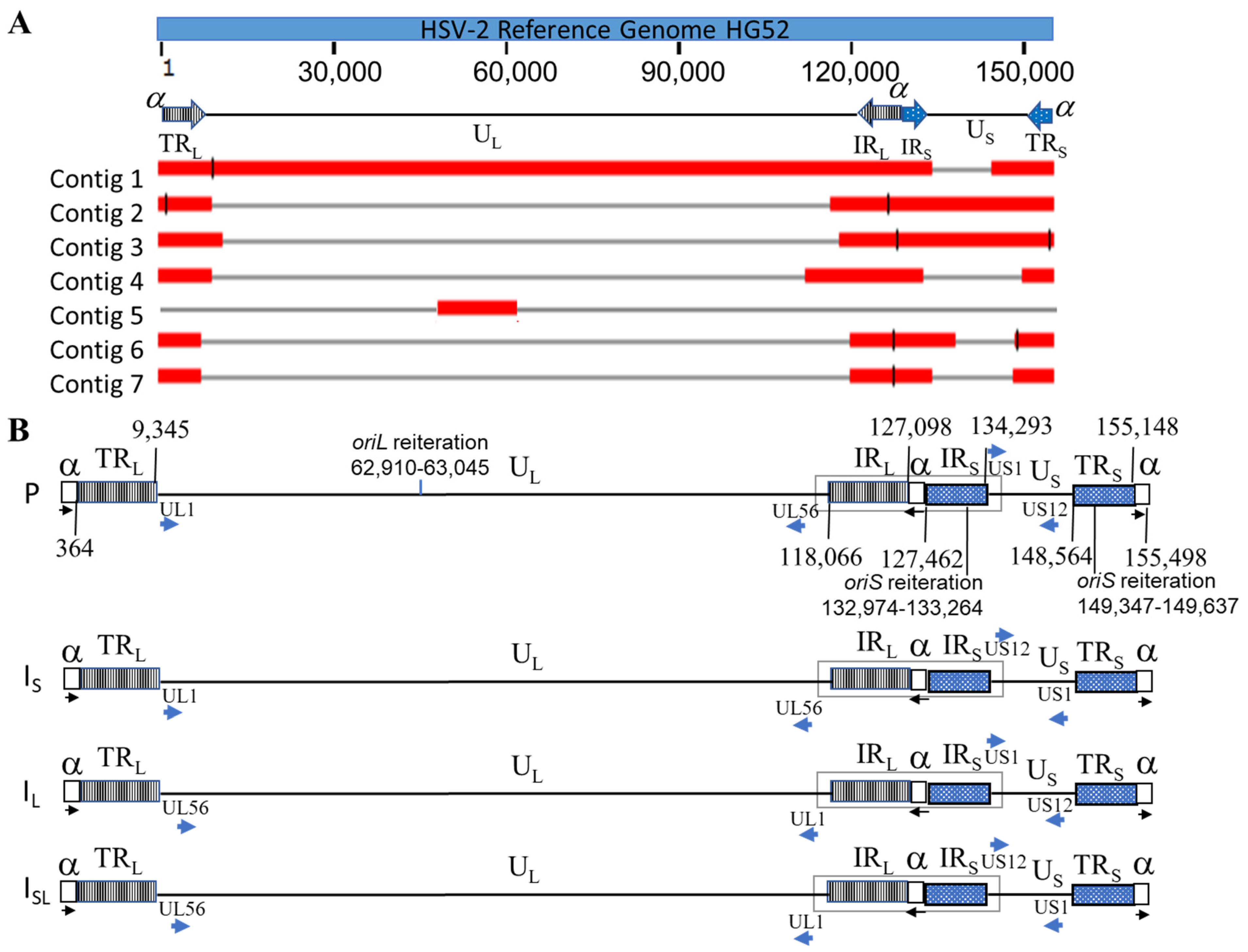
3.1. Genomic Sequencing, Assembly, and Annotation
3.2. Confirmation of Four Isomers with ONT Long Reads
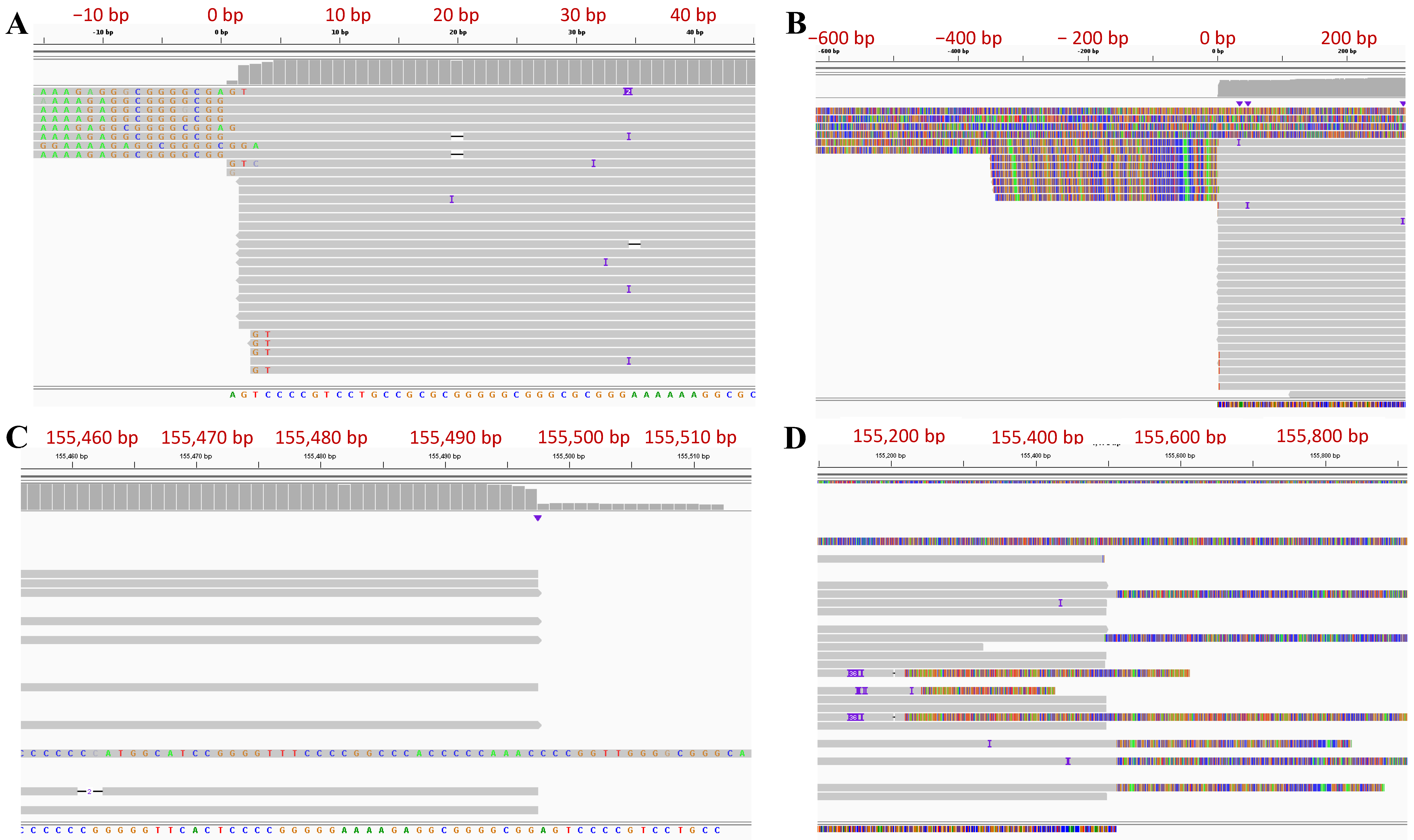
3.3. Determination of Genome Termini of HSV-2 G
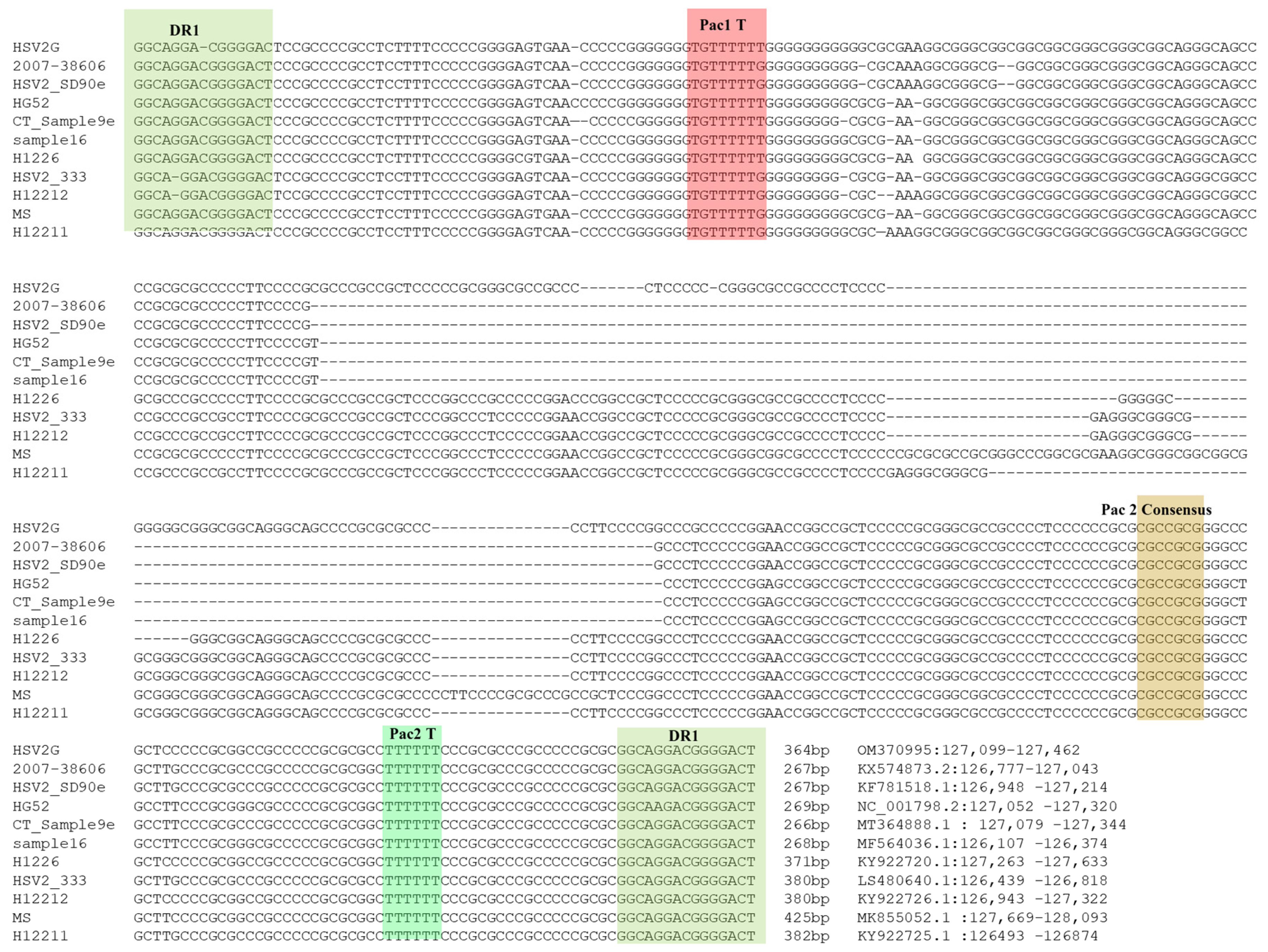
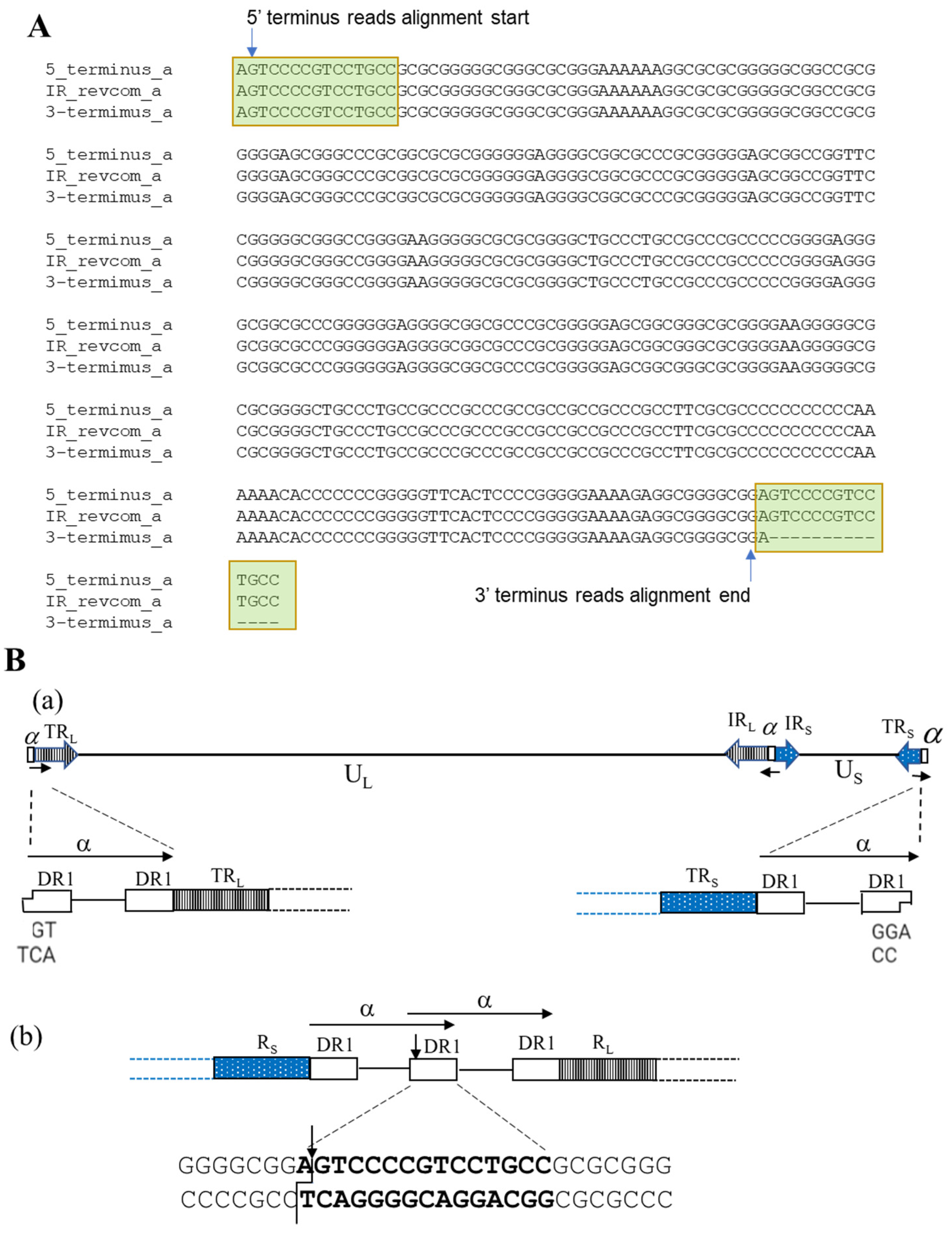
3.4. Analysis of ‘α’ Sequences of HSV2 Strain G
3.5. Determination of ‘α’ Sequence Copy Number in HSV2 Strain G Genome
3.6. Comparison of Genome Sequences between HSV-2 Strains and Phylogenetic Analysis of HSV2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitley, R.J.; Kimberlin, D.W.; Roizman, B. Herpes simplex viruses. Clin. Infect. Dis. 1998, 26, 541–555. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016. NCHS Data Brief 2018, 304, 1–8. [Google Scholar]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- del Mar Pujades Rodríguez, M.; Obasi, A.; Mosha, F.; Todd, J.; Brown, D.; Changalucha, J.; Mabey, D.; Ross, D.; Grosskurth, H.; Hayes, R. Herpes simplex virus type 2 infection increases HIV incidence: A prospective study in rural Tanzania. AIDS 2002, 16, 451–462. [Google Scholar] [CrossRef]
- Brown, J.M.; Wald, A.; Hubbard, A.; Rungruengthanakit, K.; Chipato, T.; Rugpao, S.; Mmiro, F.; Celentano, D.D.; Salata, R.S.; Morrison, C.S.; et al. Incident and prevalent herpes simplex virus type 2 infection increases risk of HIV acquisition among women in Uganda and Zimbabwe. AIDS 2007, 21, 1515–1523. [Google Scholar] [CrossRef]
- Sobngwi-Tambekou, J.; Taljaard, D.; Lissouba, P.; Zarca, K.; Puren, A.; Lagarde, E.; Auvert, B. Effect of HSV-2 serostatus on acquisition of HIV by young men: Results of a longitudinal study in Orange Farm, South Africa. J. Infect. Dis. 2009, 199, 958–964. [Google Scholar] [CrossRef]
- Todd, J.; Riedner, G.; Maboko, L.; Hoelscher, M.; Weiss, H.A.; Lyamuya, E.; Mabey, D.; Rusizoka, M.; Belec, L.; Hayes, R. Effect of genital herpes on cervicovaginal HIV shedding in women co-infected with HIV AND HSV-2 in Tanzania. PLoS ONE 2013, 8, e59037. [Google Scholar] [CrossRef]
- Ho, D.Y.; Enriquez, K.; Multani, A. Herpesvirus Infections Potentiated by Biologics. Infect. Dis. Clin. N. Am. 2020, 34, 311–339. [Google Scholar] [CrossRef]
- Ramchandani, M.; Selke, S.; Magaret, A.; Barnum, G.; Huang, M.W.; Corey, L.; Wald, A. Prospective cohort study showing persistent HSV-2 shedding in women with genital herpes 2 years after acquisition. Sex. Transm. Infect. 2018, 94, 568–570. [Google Scholar] [CrossRef]
- Sucato, G.; Wald, A.; Wakabayashi, E.; Vieira, J.; Corey, L. Evidence of latency and reactivation of both herpes simplex virus (HSV)-1 and HSV-2 in the genital region. J. Infect. Dis. 1998, 177, 1069–1072. [Google Scholar] [CrossRef]
- Williams, L.E.; Nesburn, A.B.; Kaufman, H.E. Experimental induction of disciform keratitis. Arch. Ophthalmol. 1965, 73, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, Y.; Guo, J.; Dai, Y.; Ye, L.; Qiu, J.; Zeng, Z.; Wu, X.; Xing, Y.; Long, X.; et al. Ex vivo 2D and 3D HSV-2 infection model using human normal vaginal epithelial cells. Oncotarget 2017, 8, 15267–15282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dudek, T.E.; Torres-Lopez, E.; Crumpacker, C.; Knipe, D.M. Evidence for differences in immunologic and pathogenesis properties of herpes simplex virus 2 strains from the United States and South Africa. J. Infect. Dis. 2011, 203, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Ando, Y.; Soushi, S.; Kurata, T.; Arao, Y. The BglII-N fragment of herpes simplex virus type 2 contains a region responsible for resistance to antiviral effects of interferon. J. Gen. Virol. 1998, 79 Pt 3, 565–572. [Google Scholar] [CrossRef]
- Spector, F.C.; Kern, E.R.; Palmer, J.; Kaiwar, R.; Cha, T.A.; Brown, P.; Spaete, R.R. Evaluation of a live attenuated recombinant virus RAV 9395 as a herpes simplex virus type 2 vaccine in guinea pigs. J. Infect. Dis. 1998, 177, 1143–1154. [Google Scholar] [CrossRef][Green Version]
- Petro, C.D.; Weinrick, B.; Khajoueinejad, N.; Burn, C.; Sellers, R.; Jacobs, W.R., Jr.; Herold, B.C. HSV-2 ΔgD elicits FcγR-effector antibodies that protect against clinical isolates. JCI Insight 2016, 1, e88529. [Google Scholar] [CrossRef]
- Boehmer, P.E.; Nimonkar, A.V. Herpes virus replication. IUBMB Life 2003, 55, 13–22. [Google Scholar] [CrossRef]
- Strang, B.L.; Stow, N.D. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 2005, 79, 12487–12494. [Google Scholar] [CrossRef]
- Garber, D.A.; Beverley, S.M.; Coen, D.M. Demonstration of circularization of herpes simplex virus DNA following infection using pulsed field gel electrophoresis. Virology 1993, 197, 459–462. [Google Scholar] [CrossRef]
- Zhang, X.; Efstathiou, S.; Simmons, A. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: Implications for viral DNA amplification strategies. Virology 1994, 202, 530–539. [Google Scholar] [CrossRef]
- Severini, A.; Scraba, D.G.; Tyrrell, D.L. Branched structures in the intracellular DNA of herpes simplex virus type 1. J. Virol. 1996, 70, 3169–3175. [Google Scholar] [CrossRef] [PubMed]
- Spaete, R.R.; Mocarski, E.S. The alpha sequence of the cytomegalovirus genome functions as a cleavage/packaging signal for herpes simplex virus defective genomes. J. Virol. 1985, 54, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.S.; Ambinder, R.; Ciufo, D.; Hayward, S.D.; LaFemina, R.L. Structural organization of human herpesvirus DNA molecules. J. Investig. Derm. 1984, 83, 29s–41s. [Google Scholar] [CrossRef]
- Jacob, R.J.; Morse, L.S.; Roizman, B. Anatomy of herpes simplex virus DNA. XII. Accumulation of head-to-tail concatemers in nuclei of infected cells and their role in the generation of the four isomeric arrangements of viral DNA. J. Virol. 1979, 29, 448–457. [Google Scholar] [CrossRef]
- Hayward, G.S.; Jacob, R.J.; Wadsworth, S.C.; Roizman, B. Anatomy of herpes simplex virus DNA: Evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc. Natl. Acad. Sci. USA 1975, 72, 4243–4247. [Google Scholar] [CrossRef]
- Poffenberger, K.L.; Tabares, E.; Roizman, B. Characterization of a viable, noninverting herpes simplex virus 1 genome derived by insertion and deletion of sequences at the junction of components L and S. Proc. Natl. Acad. Sci. USA 1983, 80, 2690–2694. [Google Scholar] [CrossRef]
- Dolan, A.; Jamieson, F.E.; Cunningham, C.; Barnett, B.C.; McGeoch, D.J. The genome sequence of herpes simplex virus type 2. J. Virol. 1998, 72, 2010–2021. [Google Scholar] [CrossRef]
- López-Muñoz, A.D.; Rastrojo, A.; Alcamí, A. Complete Genome Sequence of Herpes Simplex Virus 2 Strain 333. Microbiol. Resour. Announc. 2018, 7, e00870-18. [Google Scholar] [CrossRef]
- López-Muñoz, A.D.; Rastrojo, A.; Kropp, K.A.; Viejo-Borbolla, A.; Alcamí, A. Combination of long- and short-read sequencing fully resolves complex repeats of herpes simplex virus 2 strain MS complete genome. Microb. Genom. 2021, 7, 000586. [Google Scholar] [CrossRef]
- Colgrove, R.; Diaz, F.; Newman, R.; Saif, S.; Shea, T.; Young, S.; Henn, M.; Knipe, D.M. Genomic sequences of a low passage herpes simplex virus 2 clinical isolate and its plaque-purified derivative strain. Virology 2014, 450–451, 140–145. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Sui, H.; Lyons, C.; Tran, B.; Sherman, B.T.; Imamichi, T. Complete Genome Sequence of Herpes Simplex Virus 1 Strain MacIntyre. Microbiol. Resour. Announc. 2019, 8, e00895-19. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Sui, H.; Lyons, C.; Tran, B.; Sherman, B.T.; Imamichi, T. Complete Genome Sequence of Herpes Simplex Virus 1 Strain McKrae. Microbiol. Resour. Announc. 2019, 8, e00993-19. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Tong, L.; Stow, N.D. Analysis of herpes simplex virus type 1 DNA packaging signal mutations in the context of the viral genome. J. Virol. 2010, 84, 321–329. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.W.; Weber, P.C. The a sequence is dispensable for isomerization of the herpes simplex virus type 1 genome. J. Virol. 1996, 70, 8801–8812. [Google Scholar] [CrossRef]
- Huang, K.J.; Zemelman, B.V.; Lehman, I.R. Endonuclease G, a candidate human enzyme for the initiation of genomic inversion in herpes simplex type 1 virus. J. Biol. Chem. 2002, 277, 21071–21079. [Google Scholar] [CrossRef]
- Koelle, D.M.; Norberg, P.; Fitzgibbon, M.P.; Russell, R.M.; Greninger, A.L.; Huang, M.L.; Stensland, L.; Jing, L.; Magaret, A.S.; Diem, K.; et al. Worldwide circulation of HSV-2 × HSV-1 recombinant strains. Sci. Rep. 2017, 7, 44084. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.W.; Larsen, I.V.; Cuellar, J.A.; Brandt, C.R. Genomic, phylogenetic, and recombinational characterization of herpes simplex virus 2 strains. J. Virol. 2015, 89, 6427–6434. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.M.; Lamers, S.L.; Weiner, B.; Ray, S.C.; Colgrove, R.C.; Diaz, F.; Jing, L.; Wang, K.; Saif, S.; Young, S.; et al. Genome Sequencing and Analysis of Geographically Diverse Clinical Isolates of Herpes Simplex Virus 2. J. Virol. 2015, 89, 8219–8232. [Google Scholar] [CrossRef]
- Johnston, C.; Magaret, A.; Roychoudhury, P.; Greninger, A.L.; Cheng, A.; Diem, K.; Fitzgibbon, M.P.; Huang, M.L.; Selke, S.; Lingappa, J.R.; et al. Highly conserved intragenic HSV-2 sequences: Results from next-generation sequencing of HSV-2 U(L) and U(S) regions from genital swabs collected from 3 continents. Virology 2017, 510, 90–98. [Google Scholar] [CrossRef]
- Casto, A.M.; Huang, M.W.; Xie, H.; Jerome, K.R.; Wald, A.; Johnston, C.M.; Greninger, A.L. Herpes Simplex Virus Mistyping due to HSV-1 × HSV-2 Interspecies Recombination in Viral Gene Encoding Glycoprotein B. Viruses 2020, 12, 860. [Google Scholar] [CrossRef]
- Davison, A.J. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann. N. Y. Acad. Sci. 2011, 1230, E37–E49. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| αSeq_Read_Location | Total Reads | 1 αSeq | 2 αSeq | 3 αSeq | 4 αSeq |
|---|---|---|---|---|---|
| aSeqm-TRL | 207 | 171 | 31 | 3 | 2 |
| IRL-aSeqn-IRs | 196 | 121 | 62 | 11 | 2 |
| aSeq(349)-TRs | 145 | 145 | 0 | 0 | 0 |
| Strain | NCBI Accession | Genome Length | Substitution Only | In/Del Only | Substitution and In/Del | Frameshift |
|---|---|---|---|---|---|---|
| MS | MK855052.1 | 155,975 | 52 | 1 | 13 | 1 |
| HG2 | NC_001798.2 | 154,675 | 44 | 1 | 13 | 1 |
| 333 | LS480640.1 | 155,503 | 43 | 0 | 14 | 1 |
| Virus | Strains | Distance to G | Origin Location | NCBI Accession | Sequence Ref |
|---|---|---|---|---|---|
| HSV-2 | 1192 | 0.001889 | Wisconsin, United States | KP334095.1 | [46] |
| HSV-2 | 9335-2007-14 | 0.002367 | Seattle, WA, USA | KR135313.1 | [47] |
| HSV-2 | GSC-56 | 0.002409 | United States | KP334094.1 | [46] |
| HSV-2 | HG52 | 0.002414 | Scotland, United Kingdom | NC_001798.2 | [27] |
| HSV-2 | 44-419851 | 0.002414 | MD, USA | KR135309.1 | [47] |
| HSV-2 | 7444_1996_25809 | 0.002517 | Seattle, WA, USA | KR135314.1 | [47] |
| HSV-2 | CtSF | 0.002530 | United States | KP334097.1 | [46] |
| HSV-2 | 333 | 0.002567 | Texas, United States | LS480640.1 | [28] |
| HSV-2 | BethesdaP5 | 0.002605 | MD, USA | KR135330.1 | [47] |
| HSV-2 | SD66 | 0.002619 | Carletonville, South Africa | KR135320.1 | [47] |
| HSV-2 | MS | 0.002784 | Reykjavik, Iceland | MK855052.1 | [29] |
| HSV-2 | K39924 | 0.002797 | Rakai, Uganda | KR135305.1 | [47] |
| HSV-2 | CT_Sample9e | 0.002799 | Seattle, WA, USA | MT364888.1 | [49] |
| HSV-2 | CtSF-R | 0.002800 | United States | KP334093.1 | [46] |
| HSV-2 | COH3818 | 0.002809 | United States | KP334096.1 | [46] |
| HSV-2 | JA2 | 0.002870 | Japan | KR135323.1 | [47] |
| HSV-2 | SD90e | 0.002915 | Carletonville, South Africa | KF781518.1 | [30] |
| HSV-2 | 2007–8222 | 0.003046 | Zambia | KX574883.2 | [48] |
| HSV-2 | J32715 | 0.003062 | Rakai, Uganda | KR135315.1 | [47] |
| HSV-2 | 2007–38606 | 0.003264 | Peru | KX574873.2 | [48] |
| HSV-1 | strain_17 | 0.573289 | Scotland, United Kingdom | NC_001806.2 | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, W.; Jiao, X.; Sui, H.; Goswami, S.; Sherman, B.T.; Fromont, C.; Caravaca, J.M.; Tran, B.; Imamichi, T. Complete Genome Sequence of Herpes Simplex Virus 2 Strain G. Viruses 2022, 14, 536. https://doi.org/10.3390/v14030536
Chang W, Jiao X, Sui H, Goswami S, Sherman BT, Fromont C, Caravaca JM, Tran B, Imamichi T. Complete Genome Sequence of Herpes Simplex Virus 2 Strain G. Viruses. 2022; 14(3):536. https://doi.org/10.3390/v14030536
Chicago/Turabian StyleChang, Weizhong, Xiaoli Jiao, Hongyan Sui, Suranjana Goswami, Brad T. Sherman, Caroline Fromont, Juan Manuel Caravaca, Bao Tran, and Tomozumi Imamichi. 2022. "Complete Genome Sequence of Herpes Simplex Virus 2 Strain G" Viruses 14, no. 3: 536. https://doi.org/10.3390/v14030536
APA StyleChang, W., Jiao, X., Sui, H., Goswami, S., Sherman, B. T., Fromont, C., Caravaca, J. M., Tran, B., & Imamichi, T. (2022). Complete Genome Sequence of Herpes Simplex Virus 2 Strain G. Viruses, 14(3), 536. https://doi.org/10.3390/v14030536