
High-Throughput Platform for Detection of Neutralizing Antibodies Using Flavivirus Reporter Replicon Particles

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Stable Cell Line
2.2. Viruses
2.3. Plasmid Constructions
2.4. In Vitro Transcription and Electroporation
2.5. Preparation and Titration of Flavivirus Reporter Replicon Particles
2.6. Indirect Immunofluorescence
2.7. Western Blot Analysis
2.8. Neutralization Assays
2.9. Statistics
3. Results
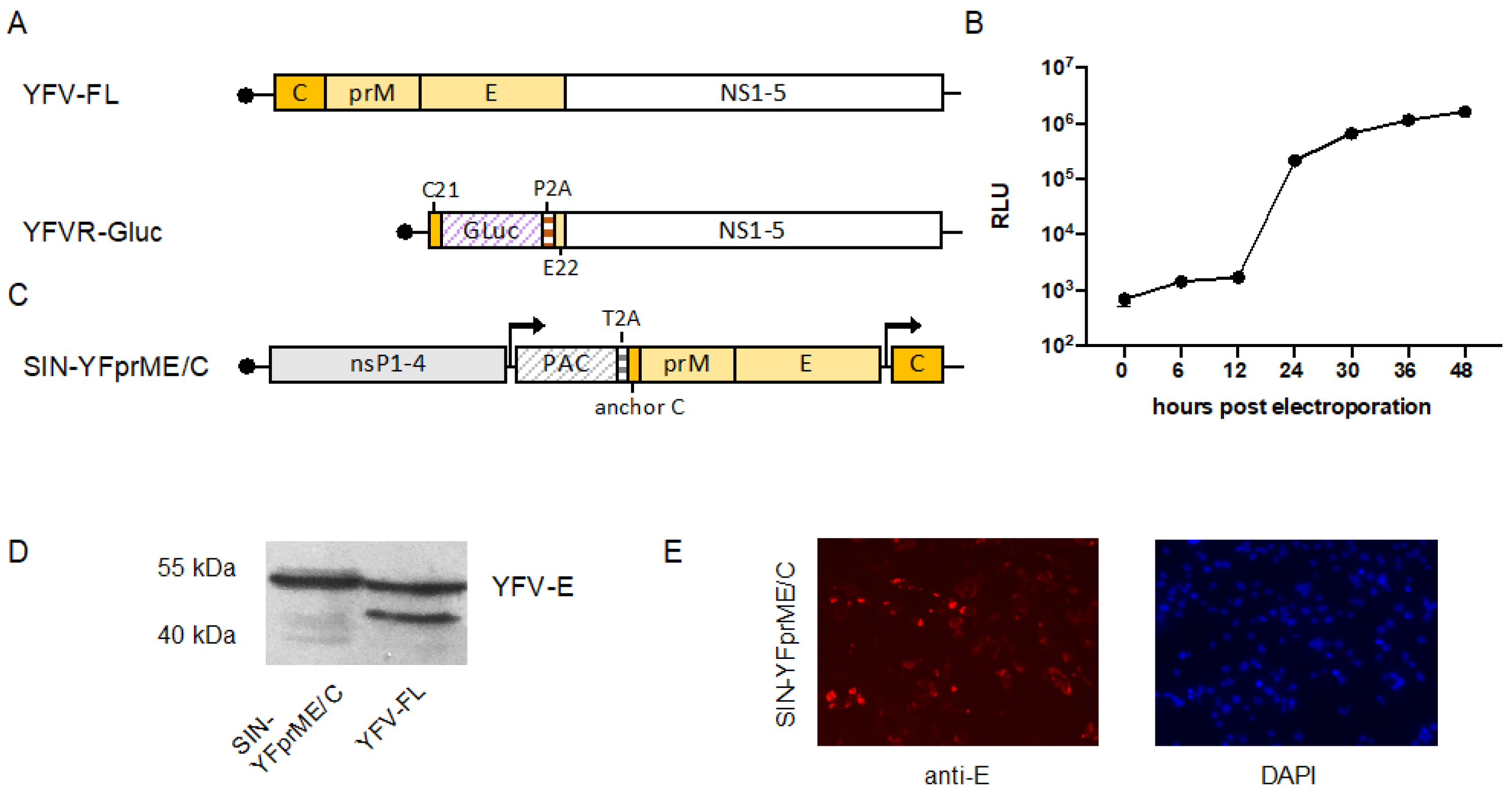
3.1. Generation of a YFV Replicon Expressing Gaussia luciferase
3.2. Establishment of a Double Subgenomic YFV Packaging Construct
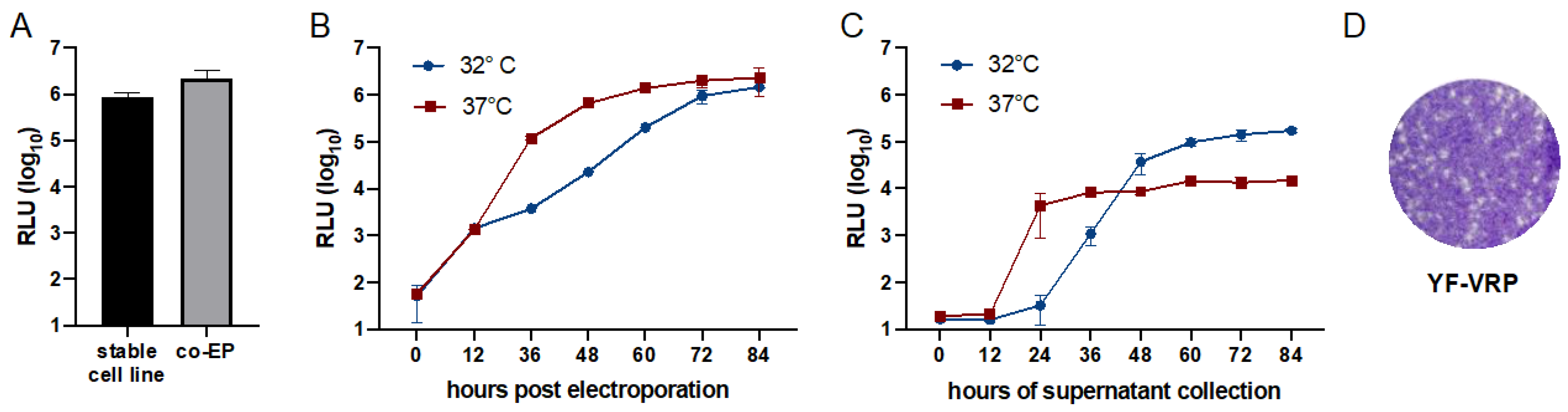
3.3. Optimization of Flavivirus VRP Production
3.4. Set-Up and Validation of YF-VRP Based Neutralization Assays
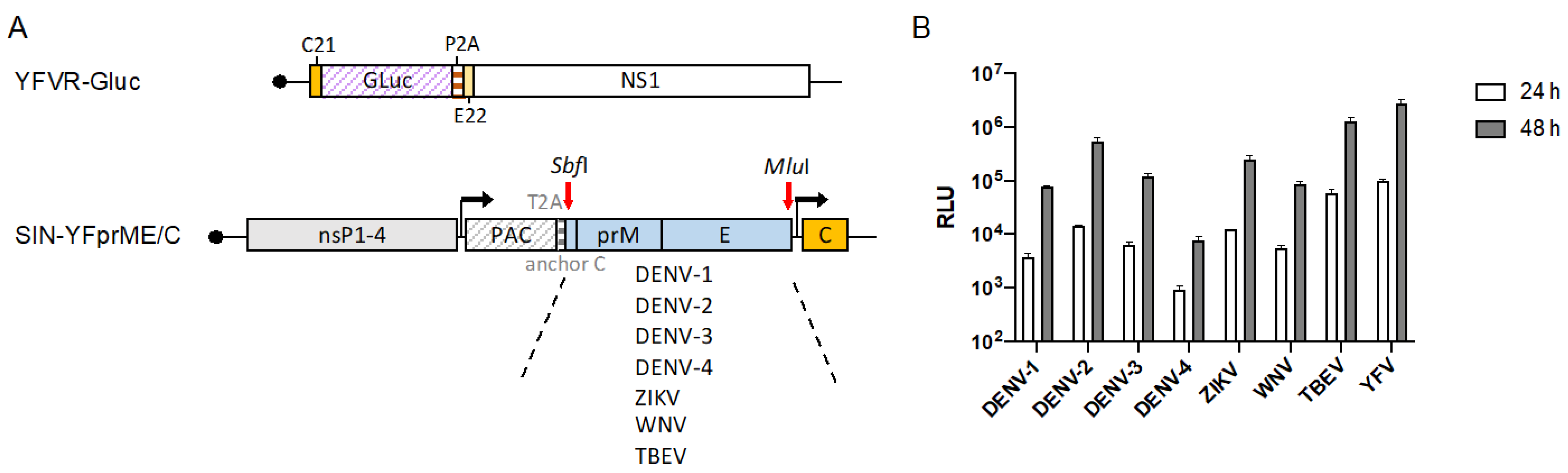
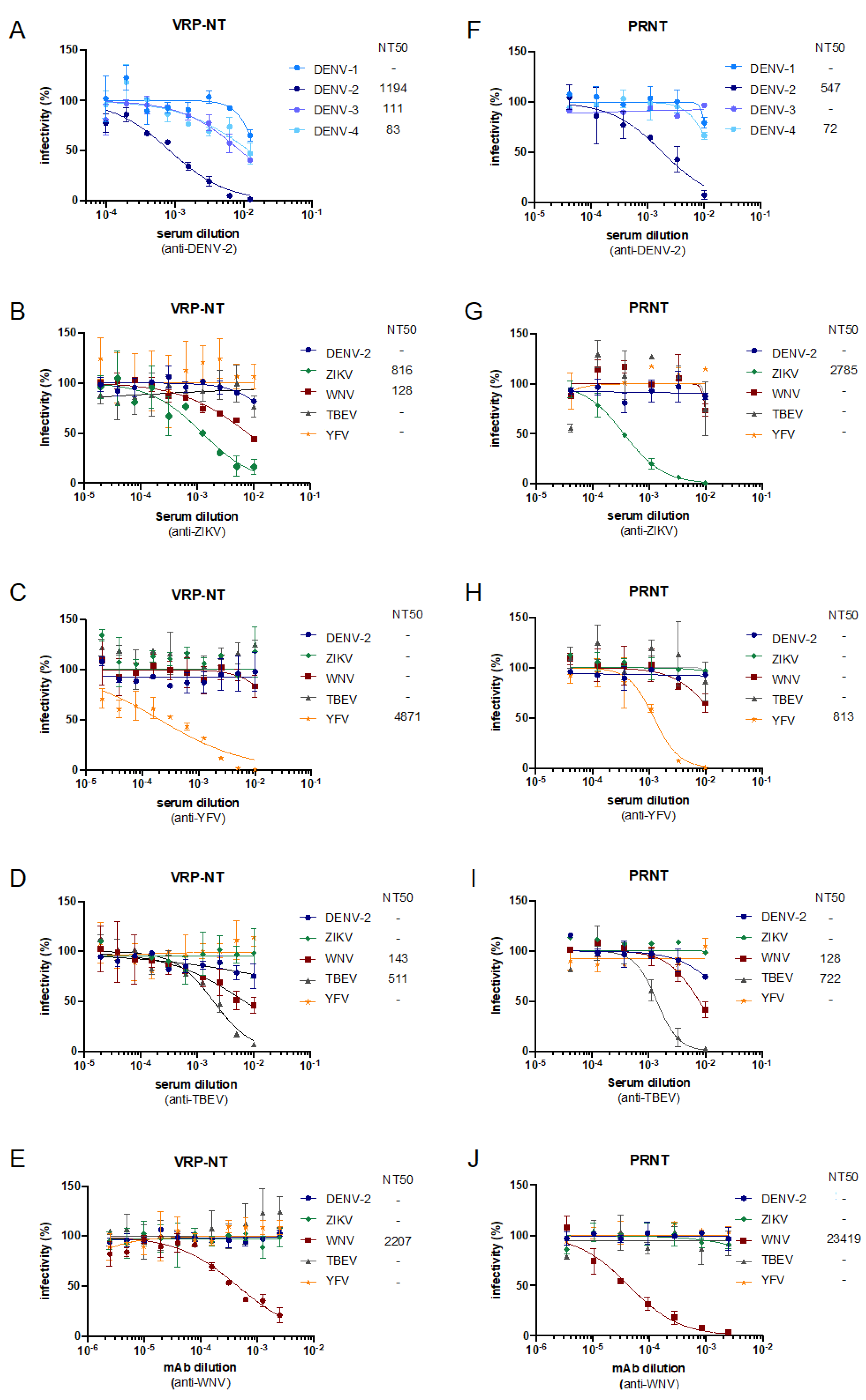
3.5. Establishment and Validation of Chimeric Flavivirus VRPs for Neutralization Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Frierson, J.G. The yellow fever vaccine: A history. Yale J. Biol. Med. 2010, 83, 77–85. [Google Scholar] [PubMed]
- Silva, N.I.O.; Sacchetto, L.; de Rezende, I.M.; Trindade, G.S.; LaBeaud, A.D.; de Thoisy, B.; Drumond, B.P. Recent sylvatic yellow fever virus transmission in Brazil: The news from an old disease. Virol. J. 2020, 17, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira Figueiredo, P.; Stoffella-Dutra, A.G.; Barbosa Costa, G.; Silva de Oliveira, J.; Dourado Amaral, C.; Duarte Santos, J.; Soares Rocha, K.L.; Araújo Júnior, J.P.; Lacerda Nogueira, M.; Zazá Borges, M.A.; et al. Re-Emergence of Yellow Fever in Brazil during 2016–2019: Challenges, Lessons Learned, and Perspectives. Viruses 2020, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- WHO. Yellow Fever—Brazil. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/11-february-2019-yellow-fever-brazil-en (accessed on 22 December 2021).
- Moreira-Soto, A.; Torres, M.C.; Lima de Mendonça, M.C.; Mares-Guia, M.A.; Dos Santos Rodrigues, C.D.; Fabri, A.A.; Dos Santos, C.C.; Machado Araújo, E.S.; Fischer, C.; Ribeiro Nogueira, R.M.; et al. Evidence for multiple sylvatic transmission cycles during the 2016–2017 yellow fever virus outbreak, Brazil. Clin. Microbiol. Infect. 2018, 24, 1019.e1–1019.e4. [Google Scholar] [CrossRef] [Green Version]
- Uchenna Emeribe, A.; Nasir Abdullahi, I.; Ajagbe, O.O.R.; Egede Ugwu, C.; Oloche Onoja, S.; Dahiru Abubakar, S.; Modesta Umeozuru, C.; Sunday Animasaun, O.; Omoruyi Omosigho, P.; Mukhtar Danmusa, U.; et al. Incidence, drivers and global health implications of the 2019/2020 yellow fever sporadic outbreaks in Sub-Saharan Africa. Pathog. Dis. 2021, 79. [Google Scholar] [CrossRef]
- Abdullahi, I.N.; Anka, A.U.; Emeribe, A.U.; Umar, K.; Adekola, H.A.; Uzairue, L.; Ghmaba, P.E.; Okwume, C.C. The interplay between environmental factors, vector competence and vaccine immunodynamics as possible explanation of the 2019 yellow fever re-emergence in Nigeria. New Microbes New Infect. 2021, 41, 100858. [Google Scholar] [CrossRef]
- Ingelbeen, B.; Weregemere, N.A.; Noel, H.; Tshapenda, G.P.; Mossoko, M.; Nsio, J.; Ronsse, A.; Ahuka-Mundeke, S.; Cohuet, S.; Kebela, B.I. Urban yellow fever outbreak-Democratic Republic of the Congo, 2016: Towards more rapid case detection. PLoS Negl. Trop. Dis. 2018, 12, e0007029. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, T.J.; Panda, P.K.; Wolford, R.W. Dengue Fever. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/28613483/ (accessed on 22 December 2021).
- Guo, C.; Zhou, Z.; Wen, Z.; Liu, Y.; Zeng, C.; Xiao, D.; Ou, M.; Han, Y.; Huang, S.; Liu, D.; et al. Global Epidemiology of Dengue Outbreaks in 1990–2015: A Systematic Review and Meta-Analysis. Front. Cell Infect. Microbiol. 2017, 7, 317. [Google Scholar] [CrossRef]
- WHO. Dengue and Severe Dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 22 December 2021).
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and Emergence of Zika Virus. J. Infect. Dis. 2017, 216, S860–S867. [Google Scholar] [CrossRef] [Green Version]
- Carod-Artal, F.J. Epidemiology and neurological complications of infection by the Zika virus: A new emerging neurotropic virus. Rev. Neurol. 2016, 62, 317–328. [Google Scholar]
- ECDC. Mosqutio Maps. Available online: https://www.ecdc.europa.eu/en/disease-vectors/surveillance-and-disease-data/mosquito-maps (accessed on 22 December 2021).
- Nash, D.; Mostashari, F.; Fine, A.; Miller, J.; O’Leary, D.; Murray, K.; Huang, A.; Rosenberg, A.; Greenberg, A.; Sherman, M.; et al. The outbreak of West Nile virus infection in the New York City area in 1999. N. Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, D.R.; Marfin, A.A.; Montgomery, S.P.; Kipp, A.M.; Lehman, J.A.; Biggerstaff, B.J.; Elko, V.L.; Collins, P.D.; Jones, J.E.; Campbell, G.L. The epidemic of West Nile virus in the United States, 2002. Vector Borne Zoonotic Dis. 2004, 4, 61–70. [Google Scholar] [CrossRef]
- Bakonyi, T.; Haussig, J.M. West Nile virus keeps on moving up in Europe. Euro Surveill. 2020, 25, 2001938. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ferenczi, E.; Erdélyi, K.; Kutasi, O.; Csörgő, T.; Seidel, B.; Weissenböck, H.; Brugger, K.; Bán, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 West Nile virus in Central Europe, 2008/2009. Vet. Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef]
- Ziegler, U.; Lühken, R.; Keller, M.; Cadar, D.; van der Grinten, E.; Michel, F.; Albrecht, K.; Eiden, M.; Rinder, M.; Lachmann, L.; et al. West Nile virus epizootic in Germany, 2018. Antivir. Res. 2019, 162, 39–43. [Google Scholar] [CrossRef]
- Riccardi, N.; Antonello, R.M.; Luzzati, R.; Zajkowska, J.; Di Bella, S.; Giacobbe, D.R. Tick-borne encephalitis in Europe: A brief update on epidemiology, diagnosis, prevention, and treatment. Eur. J. Intern. Med. 2019, 62, 1–6. [Google Scholar] [CrossRef]
- Yoshii, K.; Song, J.Y.; Park, S.B.; Yang, J.; Schmitt, H.J. Tick-borne encephalitis in Japan, Republic of Korea and China. Emerg. Microbes Infect. 2017, 6, e82. [Google Scholar]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.-J.; Rice, C.M. Flaviviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 712–746. [Google Scholar]
- Fischer, C.; Jo, W.K.; Haage, V.; Moreira-Soto, A.; de Oliveira Filho, E.F.; Drexler, J.F. Challenges towards serologic diagnostics of emerging arboviruses. Clin. Microbiol. Infect. 2021, 27, 1221–1229. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, J. Review of diagnostic plaque reduction neutralization tests for flavivirus infection. Vet. J. 2013, 195, 33–40. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Varnavski, A.N.; Westaway, E.G. Encapsidation of the flavivirus kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J. Virol. 1998, 72, 5967–5977. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, A.; Moi, M.L.; Takasaki, T.; Kurane, I.; Matsuda, M.; Suzuki, R.; Konishi, E. Utility of Japanese encephalitis virus subgenomic replicon-based single-round infectious particles as antigens in neutralization tests for Zika virus and three other flaviviruses. J. Virol. Methods 2017, 243, 164–171. [Google Scholar] [CrossRef]
- Li, W.; Ma, L.; Guo, L.P.; Wang, X.L.; Zhang, J.W.; Bu, Z.G.; Hua, R.H. West Nile virus infectious replicon particles generated using a packaging-restricted cell line is a safe reporter system. Sci. Rep. 2017, 7, 3286. [Google Scholar] [CrossRef]
- Pedroso, C.; Fischer, C.; Feldmann, M.; Sarno, M.; Luz, E.; Moreira-Soto, A.; Cabral, R.; Netto, E.M.; Brites, C.; Kümmerer, B.M.; et al. Cross-Protection of Dengue Virus Infection against Congenital Zika Syndrome, Northeastern Brazil. Emerg. Infect. Dis. 2019, 25, 1485–1493. [Google Scholar] [CrossRef] [Green Version]
- Bredenbeek, P.J.; Kooi, E.A.; Lindenbach, B.; Huijkman, N.; Rice, C.M.; Spaan, W.J.M. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J. Gen. Virol. 2003, 84, 1261–1268. [Google Scholar] [CrossRef]
- Scheck, M.K.; Lehmann, L.; Zaucha, M.; Schwarzlmueller, P.; Huber, K.; Pritsch, M.; Barba-Spaeth, G.; Thorn-Seshold, O.; Krug, A.B.; Endres, S.; et al. FluoRNT: A robust, efficient assay for detection of neutralising antibodies against yellow fever virus 17D. PLoS ONE 2022. accepted. [Google Scholar]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N1-2′O-Methylated Self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Gläsker, S.; Lulla, A.; Lulla, V.; Couderc, T.; Drexler, J.F.; Liljeström, P.; Lecuit, M.; Drosten, C.; Merits, A.; Kümmerer, B.M. Virus replicon particle based Chikungunya virus neutralization assay using Gaussia luciferase as readout. Virol. J. 2013, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Agapov, E.V.; Frolov, I.; Lindenbach, B.D.; Prágai, B.M.; Schlesinger, S.; Rice, C.M. Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 12989–12994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kümmerer, B.M.; Rice, C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002, 76, 4773–4784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinney, R.M.; Butrapet, S.; Chang, G.J.; Tsuchiya, K.R.; Roehrig, J.T.; Bhamarapravati, N.; Gubler, D.J. Construction of infectious cDNA clones for dengue 2 virus: Strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology 1997, 230, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutso, M.; Saul, S.; Rausalu, K.; Susova, O.; Žusinaite, E.; Mahalingam, S.; Merits, A. Reverse genetic system, genetically stable reporter viruses and packaged subgenomic replicon based on a Brazilian Zika virus isolate. J. Gen. Virol. 2017, 98, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Voßmann, S.; Wieseler, J.; Kerber, R.; Kümmerer, B.M. A basic cluster in the N terminus of yellow fever virus NS2A contributes to infectious particle production. J. Virol. 2015, 89, 4951–4965. [Google Scholar] [CrossRef] [Green Version]
- Hierholzer, J.C.; Killington, R.A. Virus isolation and quantitation. In Virology Methods Manual, 1st ed.; Kangro, H., Mahy, B., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; pp. 25–46. [Google Scholar]
- Tannous, B.A.; Kim, D.E.; Fernandez, J.L.; Weissleder, R.; Breakefield, X.O. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 2005, 11, 435–443. [Google Scholar] [CrossRef]
- Jones, C.T.; Patkar, C.G.; Kuhn, R.J. Construction and applications of yellow fever virus replicons. Virology 2005, 331, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 1987, 198, 33–41. [Google Scholar] [CrossRef]
- Koraka, P.; Zeller, H.; Niedrig, M.; Osterhaus, A.D.; Groen, J. Reactivity of serum samples from patients with a flavivirus infection measured by immunofluorescence assay and ELISA. Microbes Infect. 2002, 4, 1209–1215. [Google Scholar] [CrossRef]
- Nunes, J.G.C.; Nunes, B.T.D.; Shan, C.; Moraes, A.F.; Silva, T.R.; de Mendonça, M.H.R.; das Chagas, L.L.; Silva, F.A.E.; Azevedo, R.S.S.; da Silva, E.V.P.; et al. Reporter Virus Neutralization Test Evaluation for Dengue and Zika Virus Diagnosis in Flavivirus Endemic Area. Pathogens 2021, 10, 840. [Google Scholar] [CrossRef]
- Matsuda, M.; Yamanaka, A.; Yato, K.; Yoshii, K.; Watashi, K.; Aizaki, H.; Konishi, E.; Takasaki, T.; Kato, T.; Muramatsu, M.; et al. High-throughput neutralization assay for multiple flaviviruses based on single-round infectious particles using dengue virus type 1 reporter replicon. Sci. Rep. 2018, 8, 16624. [Google Scholar] [CrossRef]
- Yamanaka, A.; Suzuki, R.; Konishi, E. Evaluation of single-round infectious, chimeric dengue type 1 virus as an antigen for dengue functional antibody assays. Vaccine 2014, 32, 4289–4295. [Google Scholar] [CrossRef]
- Harvey, T.J.; Liu, W.J.; Wang, X.J.; Linedale, R.; Jacobs, M.; Davidson, A.; Le, T.T.; Anraku, I.; Suhrbier, A.; Shi, P.Y. Tetracycline-inducible packaging cell line for production of flavivirus replicon particles. J. Virol. 2004, 78, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Amberg, S.M.; Rice, C.M. Mutagenesis of the NS2B-NS3-mediated cleavage site in the flavivirus capsid protein demonstrates a requirement for coordinated processing. J. Virol. 1999, 73, 8083–8094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritoh, K.; Maeda, A.; Nishino, T.; Sasaki, N.; Agui, T. Development and application of West Nile virus subgenomic replicon RNA expressing secreted alkaline phosphatase. J. Vet. Med. Sci. 2011, 73, 683–686. [Google Scholar] [CrossRef] [Green Version]
- Tannous, B.A. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 2009, 4, 582–591. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lücke, A.-C.; vom Hemdt, A.; Wieseler, J.; Fischer, C.; Feldmann, M.; Rothenfusser, S.; Drexler, J.F.; Kümmerer, B.M. High-Throughput Platform for Detection of Neutralizing Antibodies Using Flavivirus Reporter Replicon Particles. Viruses 2022, 14, 346. https://doi.org/10.3390/v14020346
Lücke A-C, vom Hemdt A, Wieseler J, Fischer C, Feldmann M, Rothenfusser S, Drexler JF, Kümmerer BM. High-Throughput Platform for Detection of Neutralizing Antibodies Using Flavivirus Reporter Replicon Particles. Viruses. 2022; 14(2):346. https://doi.org/10.3390/v14020346
Chicago/Turabian StyleLücke, Arlen-Celina, Anja vom Hemdt, Janett Wieseler, Carlo Fischer, Marie Feldmann, Simon Rothenfusser, Jan Felix Drexler, and Beate Mareike Kümmerer. 2022. "High-Throughput Platform for Detection of Neutralizing Antibodies Using Flavivirus Reporter Replicon Particles" Viruses 14, no. 2: 346. https://doi.org/10.3390/v14020346
APA StyleLücke, A.-C., vom Hemdt, A., Wieseler, J., Fischer, C., Feldmann, M., Rothenfusser, S., Drexler, J. F., & Kümmerer, B. M. (2022). High-Throughput Platform for Detection of Neutralizing Antibodies Using Flavivirus Reporter Replicon Particles. Viruses, 14(2), 346. https://doi.org/10.3390/v14020346