
One-Enzyme RTX-PCR for the Detection of RNA Viruses from Multiple Virus Genera and Crop Plants


 and
and 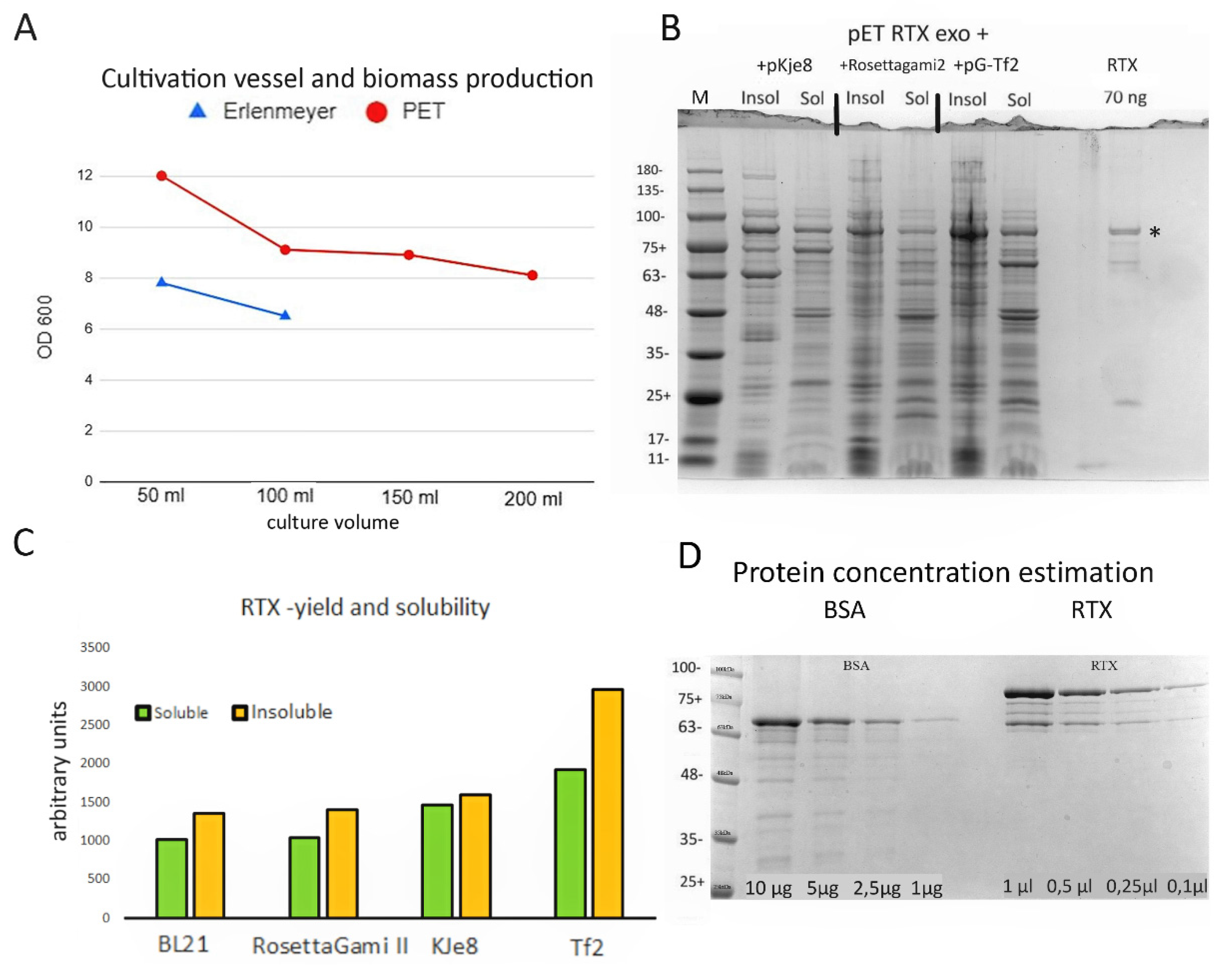{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Expression and Purification of Recombinant RTX
2.2. Virus Sources
2.3. Sample Preparation for Virus Detection (RNA Isolation, Virus Purification, and Crude Sap Preparation)
2.3.1. Virus Purification
2.3.2. RNA Isolation
2.3.3. Crude Sap Preparation
2.4. One-Step RT-PCR with RTX and Commercial Qiagen One-Step Kit
2.4.1. One-Enzyme RTX-PCR
2.4.2. One-Step RT-PCR with the Kit
3. Results and Discussion
3.1. RTX Enzyme Purification
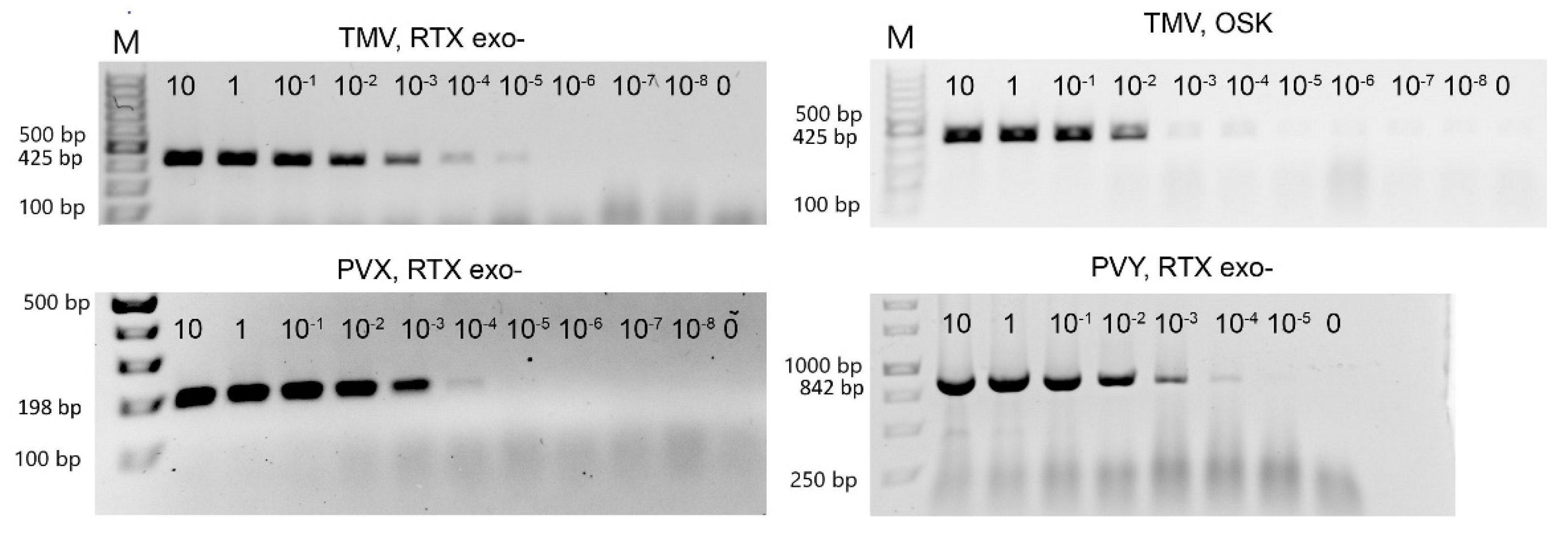
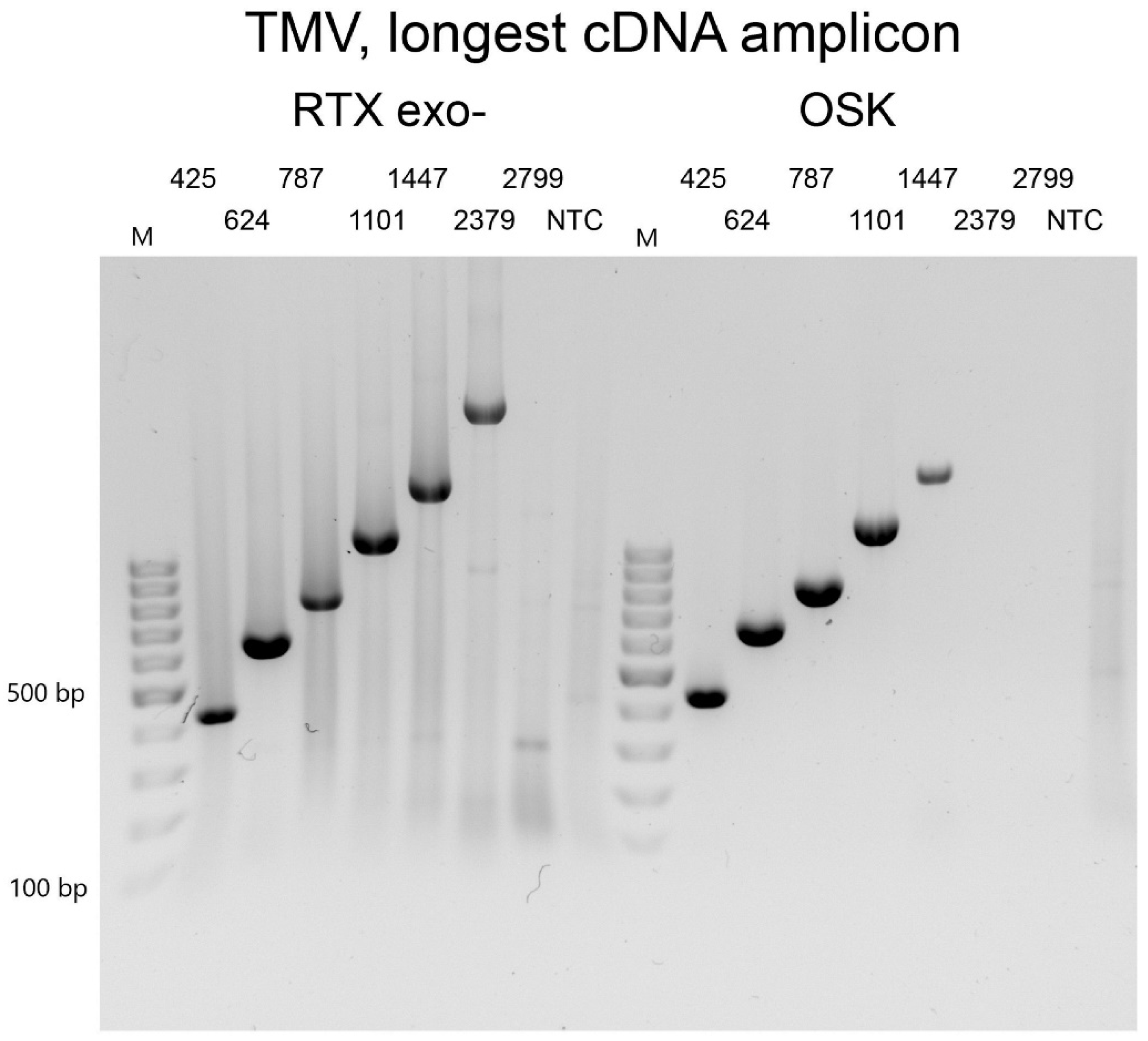
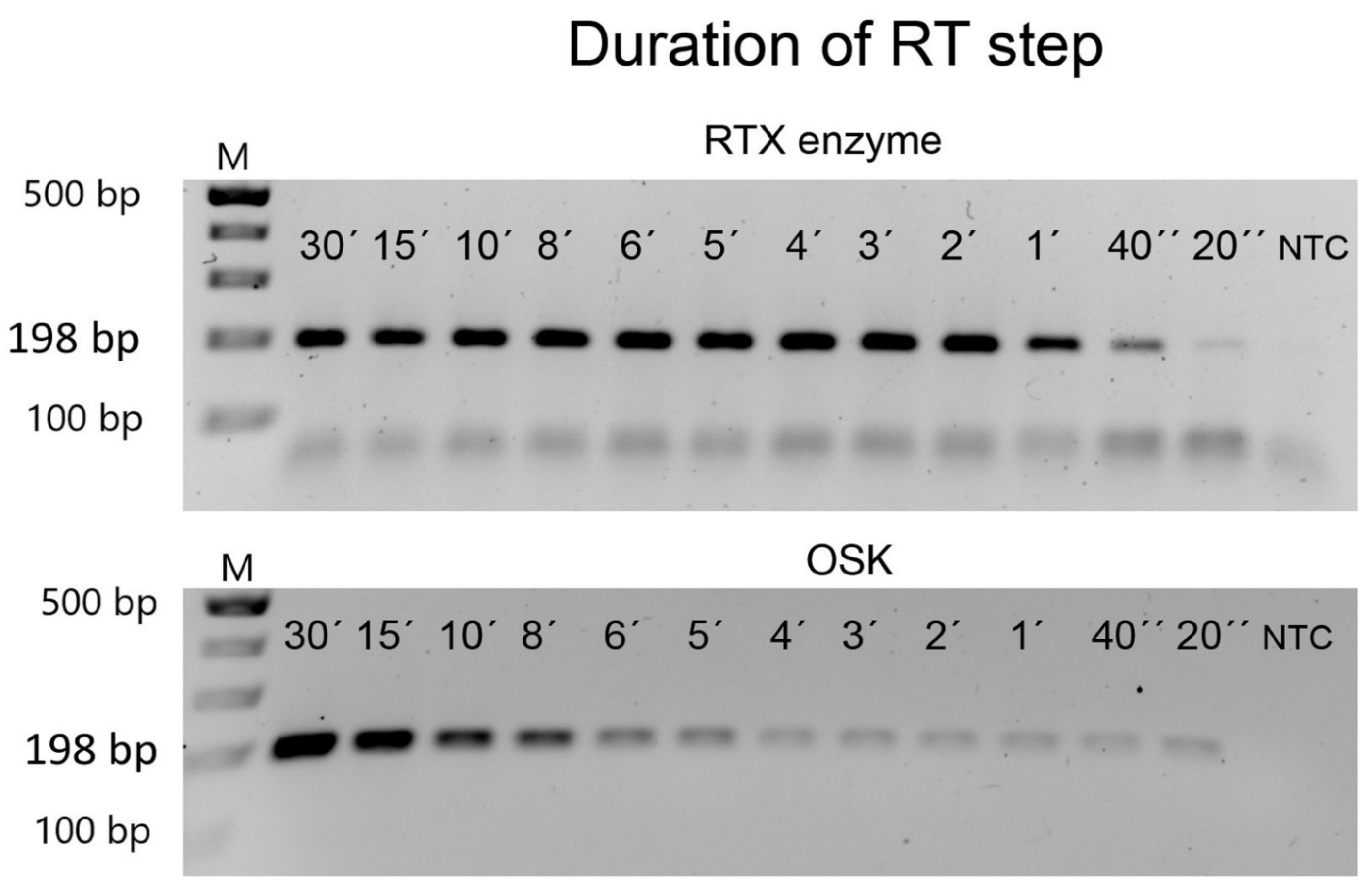
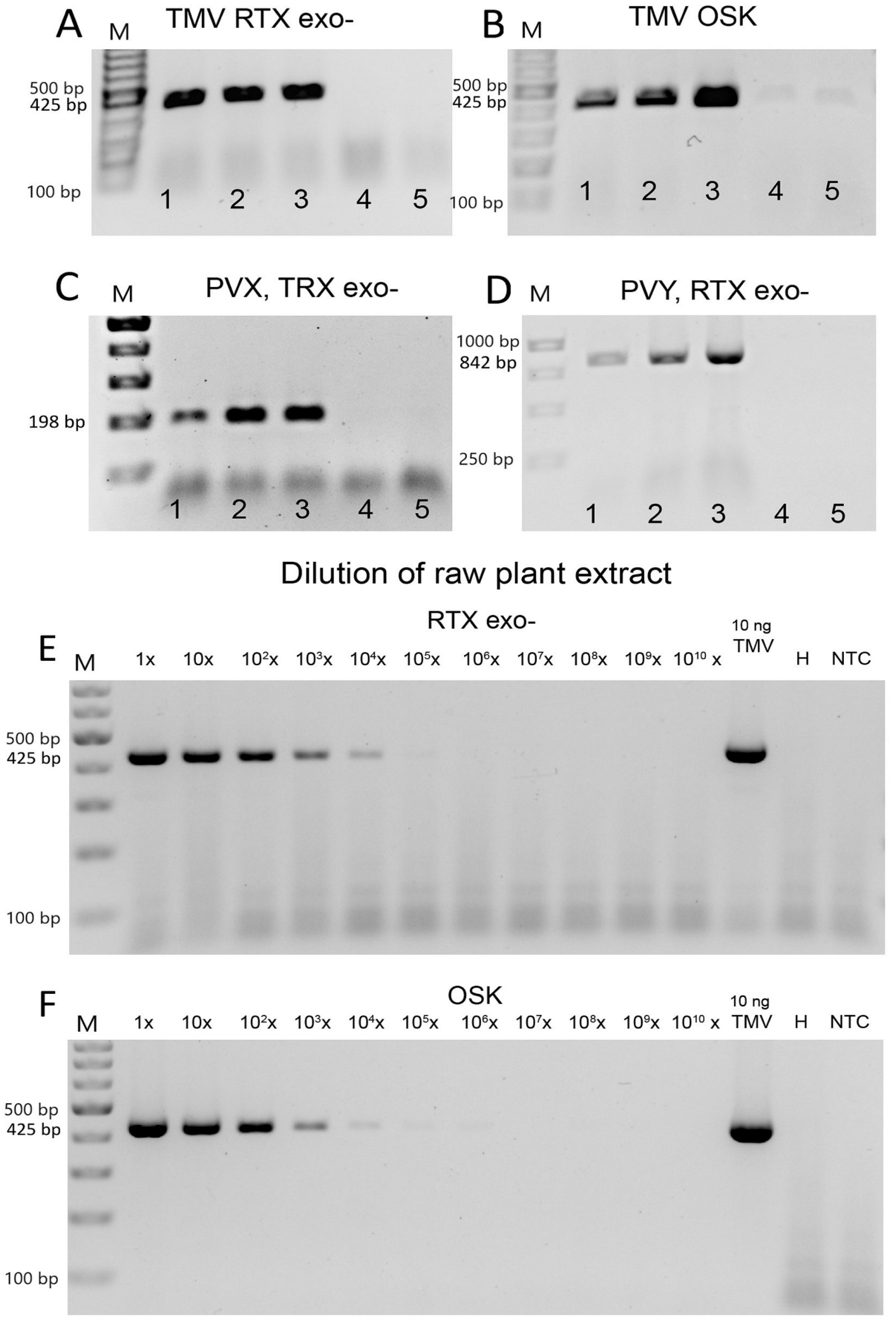
3.2. Efficiency of Virus Detection with Purified Viruses
3.3. Virus Detection Based on One-Enzyme RTX-PCR Using Crude Plant Sap
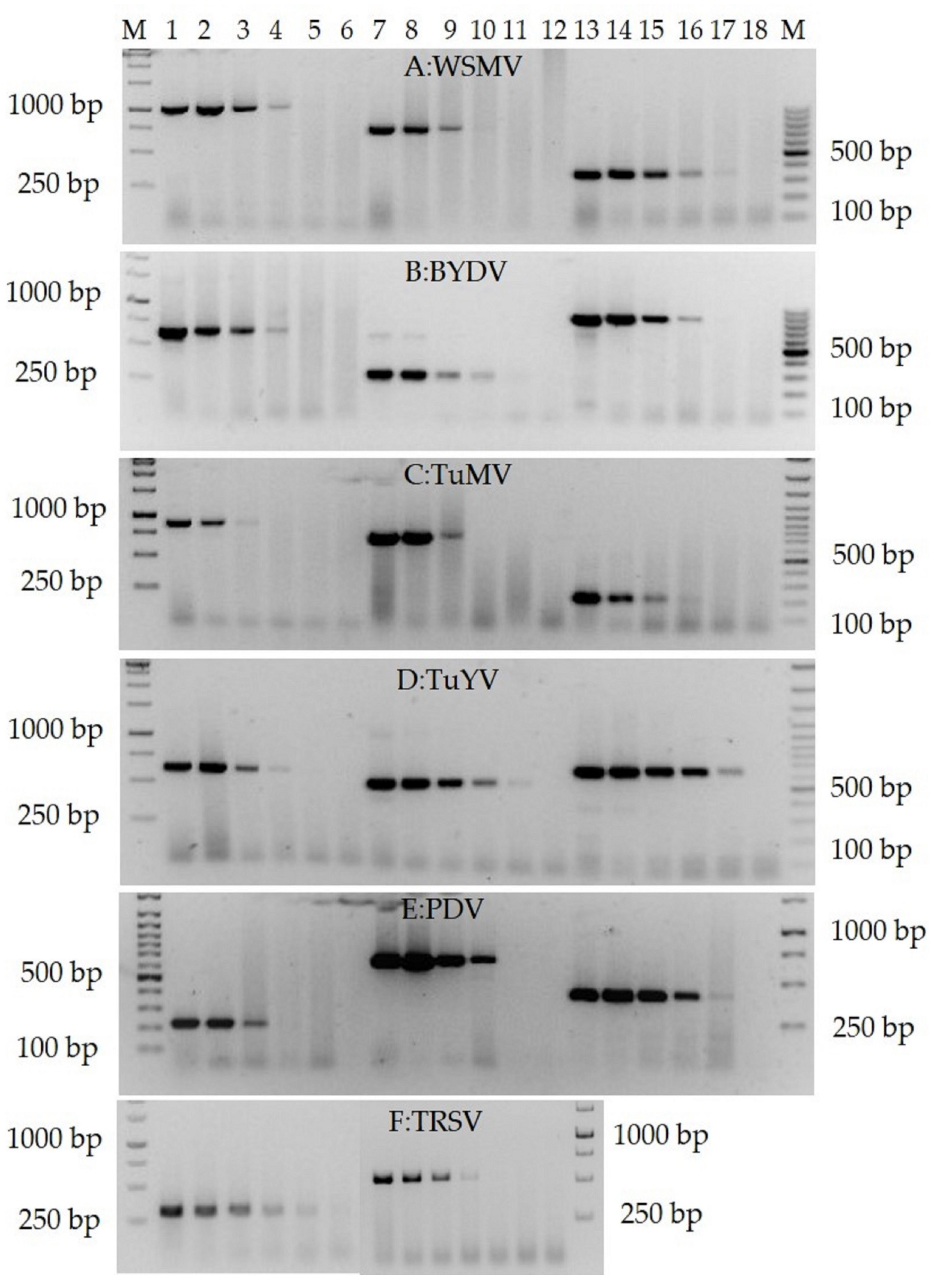
3.4. Validation of the RTX-PCR Assay for the Detection of Virus Species from Different Genera and Crops
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, R.A.C. Plant virus emergence and evolution: Origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Res. 2009, 141, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Mumford, R.A.; Macarthur, R.; Boonham, N. The role and challenges of new diagnostic technology in plant biosecurity. Food Sec. 2016, 8, 103–109. [Google Scholar] [CrossRef]
- Rubio, L.; Galiplenso, L.; Ferriol, I. Detection of plant viruses and disease management: Relevance of genetic diversity and evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Scalenghe, R.; Davino, S.; Panno, S.; Scuderi, G.; Ruisi, P.; Villa, P.; Stroppiana, D.; Boschetti, M.; Goulart, L.R.; et al. Advanced methods of plant disease detection. A review. Agron. Sustain. Dev. 2015, 35, 1–25. [Google Scholar] [CrossRef]
- Pallás, V.; Sánchez-Navarro, J.A.; James, D. Recent advances on the multiplex molecular detection of plant viruses and viroids. Front. Microbiol. 2018, 9, 2087. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, K.; Bardani, E.; Kallemi, P.; Kalantidis, K. Viral detection: Past, present, and future. BioEssays 2019, 41, 1900049. [Google Scholar] [CrossRef]
- Yasukawai, K.; Yanagihara, I.; Fujiwara, S. Alteration of enzymes and their application to nucleic acid amplification (Review). Int. J. Mol. Med. 2020, 46, 1633–1643. [Google Scholar] [CrossRef]
- Ellefson, J.W.; Gollihar, J.; Shroff, R.; Shivram, H.; Iyer, V.R.; Ellington, A.D. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science 2016, 352, 1590–1593. [Google Scholar] [CrossRef]
- Okano, H.; Baba, M.; Hidese, R.; Iida, K.; Li, T.; Kojima, K.; Takita, T.; Yanagihara, I.; Fujiwara, S.; Yasukawa, K. Accurate fidelity analysis of the reverse transcriptase by a modified next-generation sequencing. Enzyme Microb. Technol. 2018, 115, 81–85. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Preparation and transformation of competent E. coli using calcium chloride. Cold Spring Harb. Protoc. 2006, 1, 3932. [Google Scholar] [CrossRef]
- Taylor, T.; Denson, J.-P.; Esposito, D. Optimizing expression and solubility of proteins in E. coli using modified media and induction parameters. In Heterologous Gene Expression in E. coli; Humana Press: New York, NY, USA, 2017; pp. 65–82. [Google Scholar]
- Chen, S.; Zheng, X.; Cao, H.; Jiang, L.; Liu, F.; Sun, X. A simple and efficient method for extraction of Taq DNA polymerase. Electr. J. Biotechnol. 2015, 18, 355–358. [Google Scholar] [CrossRef][Green Version]
- Bruening, G.; Beachy, R.N.; Scalla, R.; Zaitlin, M. In vitro and in vivo translation of the ribonucleic acid of a cowpea strain of tobacco mosaic virus. Virology 1976, 71, 498–517. [Google Scholar] [CrossRef]
- Čeřovská, N.; Filigarová, M.; Branišová, H.; Žák, P.; Dědič, P. Some factors influencing purification of Potato virus A (PVA). Acta Virol. 1991, 35, 469–471. [Google Scholar]
- Janda, M.; Navrátil, O.; Haisel, D.; Jindřichová, B.; Fousek, J.; Burketová, L.; Čeřovská, N.; Moravec, T. Growth and stress response in Arabidopsis thaliana, Nicotiana benthamiana, Glycine max, Solanum tuberosum and Brassica napus cultivated under polychromatic LEDs. Plant Methods 2015, 11, 31. [Google Scholar] [CrossRef]
- Association of Applied Biologists. Description of Plant Viruses (PVY, PVX a TMV). Available online: https://www.dpvweb.net/ (accessed on 24 January 2022).
- Moravec, T.; Čeřovská, N.; Boonham, N. The detection of recombinant, tuber necrosing isolates of Potato virus Y (PVY(NTN)) using a three-primer PCR based in the coat protein gene. J. Virol. Methods 2003, 109, 63–68. [Google Scholar] [CrossRef]
- Fuchs, M.; Abawi, G.S.; Marsella-Herrick, P.; Cox, R.; Cox, K.D.; Carroll, J.E.; Martin, R.R. Occurrence of Tomato ringspot virus and Tobacco ringspot virus in highbush blueberry in New York state. J. Plant Pathol. 2010, 92, 451–459. [Google Scholar]
- Yang, W.; Yun, Z.; Chen., Z.; Zhang, G.; Wu, S. Detection of Tobacco ringspot virus by RT-real-time PCR. Acta Phytopathol. Sin. 2007, 34, 157–160. [Google Scholar]
- Jossey, S.; Babadoost, M. First report of Tobacco ringspot virus in pumpkin (Cucurbita pepo) in Illinois. Plant Dis. 2006, 90, 1361. [Google Scholar] [CrossRef]
- Singh, K.; Slavíková, L.; Kumar, J. Reakční směs pro detekci viru mozaiky vodnice v nekulturních a plevelných hostitelích pomocí RT-PCR (Reaction mixture for the detection of Turnip mosaic virus in non-cultural and weed hosts by RT-PCR.). Util. Model 2020, 34520. [Google Scholar]
- Abraham, A.D.; Menzel, W.; Lesemann, D.-E.; Varrelmann, M.; Vetten, H.J. Chickpea chlorotic stunt virus: A new polerovirus infecting cool-season food legumes in Ethiopia. Phytopathology 2006, 96, 437–446. [Google Scholar] [CrossRef]
- Singh, K.; Slavíková, L.; Kumar, J. Reakční směs pro detekci viru žloutenky vodnice v nekulturních a plevelných hostitelích pomocí RT-PCR (Reaction mixture for the detection of Turnip yellows virus in non-cultural and weed hosts by RT-PCR). Util. Model 2021, 35270. [Google Scholar]
- Kundu, J.K. First Report of barley yellow dwarf virus-PAS in wheat and barley grown in the Czech Republic. Plant Dis. 2008, 92, 1587. [Google Scholar] [CrossRef] [PubMed]
- Jarošová, J.; Kundu, J.K. Validation of reference genes as internal control for studying viral infections in cereals by quantitative real-time RT-PCR. BMC Plant Biol. 2010, 10, 146. [Google Scholar] [CrossRef]
- Singh, K.; Kundu, J.K. Variations in Wheat streak mosaic virus coat protein sequence among crop and non-crop hosts. Crop Pasture Sci. 2017, 68, 328–336. [Google Scholar] [CrossRef]
- Kúdela, O.; Kúdelová, M.; Nováková, S.; Glasa, M. First report of Wheat streak mosaic virus in Slovakia. Plant Dis. 2008, 92, 1365. [Google Scholar] [CrossRef] [PubMed]
- Gadiou, S.; Kudela, O.; Ripl, J.; Rabenstein, F.; Kundu, J.K.; Glasa, M. An amino acid deletion in Wheat streak mosaic virus capsid protein distinguishes a homogeneous group of European isolates and facilitates their specific detection. Plant Dis. 2009, 93, 1209–1213. [Google Scholar] [CrossRef]
- Jelkmann, W.; Keim-Konrad, R. Immuno-capture polymerase chain reaction and plate-trapped ELISA for the detection of Apple stem pitting virus. J. Phytopathol. 1997, 145, 499–503. [Google Scholar] [CrossRef]
- Gadiou, S.; Kundu, J.K.; Paunovic, S.; Garcia-Diez, P.; Komorowska, B.; Gospodaryk, A.; Handa, A.; Massart, S.; Birisik, N.; Takur, P.D.; et al. Genetic diversity of flexiviruses infecting pome fruit trees. J. Plant Pathol. 2010, 92, 687–693. [Google Scholar]
- Menzel, W.; Jelkmann, W.; Maiss, E. Detection of four apple viruses by multiplex RT-PCR assays with coamplification of plant mRNA as internal control. J. Virol. Methods 2002, 99, 81–92. [Google Scholar] [CrossRef]
- James, D. A simple and reliable protocol for the detection of Apple stem grooving virus by RT-PCR and in a multiplex PCR assay. J. Virol. Methods 1999, 83, 1–9. [Google Scholar] [CrossRef]
- Varga, A.; James, D. Detection and differentiation of Plum pox virus using real time multiplex PCR with SYBR Green and melting curve analysis: A rapid method for strain typing. J. Virol. Methods 2005, 123, 213–220. [Google Scholar] [CrossRef]
- Šubr, Z.; Pittnerova, S.; Glasa, M. A simplified RT-PCR-based detection of recombinant Plum pox virus isolates. Acta Virol. 2004, 48, 173–176. [Google Scholar]
- Jarošová, J.; Kundu, J.K. Simultaneous detection of stone fruit tree viruses by one-step multiplex RT-PCR. Sci. Hortic. 2010, 125, 68–72. [Google Scholar] [CrossRef]
- Massart, S.; Brostaux, Y.; Barbarossa, L.; César, V.; Cieslinska, M.; Dutrecq, O.; Fonseca, F.; Guillem, R.; Laviña, A.; Olmos, A.; et al. Inter-laboratory evaluation of a duplex RT-PCR method using crude extracts for the simultaneous detection of Prune dwarf virus and Prunus necrotic ringspot virus. Eur. J. Plant Pathol. 2008, 122, 539–547. [Google Scholar] [CrossRef]
- Jarošová, J.; Kundu, J.K. Detection of Prune dwarf virus by one-step RT-PCR and its quantification by real-time PCR. J. Virol. Methods 2010, 164, 139–144. [Google Scholar] [CrossRef]
- Dušek, J.; Plchová, H.; Čeřovská, N.; Poborilova, Z.; Navrátil, O.; Kratochvílová, K.; Gunter, C.; Jacobs, R.; Hitzeroth, I.I.; Rybicki, E.P.; et al. Extended set of GoldenBraid compatible vectors for fast assembly of multigenic constructs and their use to create geminiviral expression vectors. Front. Plant Sci. 2020, 11, 522059. [Google Scholar] [CrossRef]
- Bhadra, S.; Maranhao, A.C.; Paik, I.; Ellington, D.A. A one-enzyme RT-qPCR assay for SARS-CoV-2, and procedures for reagent production. Bio-Protoc. 2021, 11, e3898. [Google Scholar] [CrossRef]
- Gawande, S.J.; Shukla, A.; Chimote, V.P.; Kaushal, N.; Kaundal, P.; Garg, I.D.; Chimote, K.P. Development of PCR-based techniques for the detection of immobilised Potato virus Y virions. J. Plant Pathol. 2011, 93, 127–132. [Google Scholar]
- Peiró, A.; Pallás, V.; Sánchez-Navarro, J.Á. Simultaneous detection of eight viruses and two viroids affecting stone fruit trees by using a unique polyprobe. Eur. J. Plant Pathol. 2012, 132, 469–475. [Google Scholar] [CrossRef]
- Uga, H.; Tsuda, S. A one–step reverse transcription–polymerase chain reaction system for the simultaneous detection and identification of multiple tospovirus infections. Phytopathology 2005, 95, 166–171. [Google Scholar] [CrossRef]
- Kundu, J.K. A rapid and effective RNA release procedure for virus detection in woody plants by reverse transcription-polymerase chain reaction. Acta Virol. 2003, 47, 147–151. [Google Scholar]
- Kundu, J.K.; Rysanek, P. Detection of Beet yellows virus by RT-PCR and immunocapture RT-PCR in Tetragonia expansa and Beta vulgaris. Acta Virol. 2004, 48, 177–182. [Google Scholar]
- Thomson, D.; Dietzgen, G.R. Detection of DNA and RNA plant viruses by PCR and RT-PCR using a rapid virus release protocol without tissue homogenization. J. Virol. Methods 1995, 54, 85–95. [Google Scholar] [CrossRef]
- French, R.; Robertson, N.L. Simplified sample preparation for detection of Wheat streak mosaic virus and Barley yellow dwarf virus by PCR. J. Virol. Methods 1994, 49, 93–99. [Google Scholar] [CrossRef]
- Wetzel, T.; Candresse, T.; Macquaire, G.; Ravelonandro, M.; Dunez, J. A highly sensitive immunocapture polymerase chain reaction method for plum pox potyvirus detection. J. Virol. Methods 1992, 39, 27–37. [Google Scholar] [CrossRef]
- Fenby, N.S.; Scott, N.W.; Slater, A.; Elliott, M.C. PCR and non-isotopic labeling techniques for plant virus detection. Cell. Mol. Biol. 1995, 41, 639–652. [Google Scholar]
- Zhang, S.; Ravelonandro, M.; Russell, P.; McOwen, N.; Briard, P.; Bohannon, S.; Vrient, A. Rapid diagnostic detection of Plum pox virus in Prunus plants by isothermal AmplifyRP® using reverse transcription-recombinase polymerase amplification. J. Virol. Methods 2014, 207, 114–120. [Google Scholar] [CrossRef]
- Wilisiani, F.; Tomiyama, A.; Katoh, H.; Hartono, S.; Neriya, Y.; Nishigawa, H.; Natsuaki, T. Development of a LAMP assay with a portable device for real-time detection of begomoviruses under field conditions. J. Virol. Methods 2019, 265, 71–76. [Google Scholar] [CrossRef]
- Drygin, Y.F.; Blintsov, A.N.; Grigorenko, V.G.; Andreeva, I.P.; Osipov, A.P.; Varitzev, Y.A.; Uskov, A.I.; Kravchenko, D.V.; Atabekov, J.G. Highly sensitive field test lateral flow immunodiagnostics of PVX infection. Appl. Microbiol. Biotechnol. 2012, 93, 179–189. [Google Scholar] [CrossRef]
- Koczula, M.K.; Gallotta, A. Lateral flow assays. Essays Biochem. 2016, 60, 111–120. [Google Scholar]
- Safenkova, V.I.; Pankratova, K.G.; Zaitsev, A.I.; Varitsev, A.Y.; Vengerov, Y.Y.; Zherdev, V.A.; Dzantiev, B.B. Multiarray on a test strip (MATS): Rapid multiplex immunodetection of priority potato pathogens. Anal. Bioanal. Chem. 2016, 408, 6009–6017. [Google Scholar] [CrossRef]
- Rohrman, B.A.; Leautaud, V.; Molyneux, E.; Richards-Kortum, R.R. A Lateral Flow Assay for quantitative detection of amplified HIV-1 RNA. PLoS ONE 2012, 7, e45611. [Google Scholar] [CrossRef]
- Jailani, K.A.A.; Hendricks, K.; Roberts, D.P.; Paret, L.M. Development of a simple one-step multiplex RT-PCR system for simultaneous detection of DNA and RNA viruses of Cucurbit leaf crumple virus, Cucurbit yellow stunting disorder virus, Squash vein yellowing virus, and Cucurbit chlorotic yellows virus. Physiol. Mol. Plant Pathol. 2021, 116, 101734. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmeisterová, H.; Kratochvílová, K.; Čeřovská, N.; Slavíková, L.; Dušek, J.; Muller, K.; Fousek, J.; Plchová, H.; Navrátil, O.; Kundu, J.K.; et al. One-Enzyme RTX-PCR for the Detection of RNA Viruses from Multiple Virus Genera and Crop Plants. Viruses 2022, 14, 298. https://doi.org/10.3390/v14020298
Hoffmeisterová H, Kratochvílová K, Čeřovská N, Slavíková L, Dušek J, Muller K, Fousek J, Plchová H, Navrátil O, Kundu JK, et al. One-Enzyme RTX-PCR for the Detection of RNA Viruses from Multiple Virus Genera and Crop Plants. Viruses. 2022; 14(2):298. https://doi.org/10.3390/v14020298
Chicago/Turabian StyleHoffmeisterová, Hana, Kateřina Kratochvílová, Noemi Čeřovská, Lucie Slavíková, Jakub Dušek, Karel Muller, Jan Fousek, Helena Plchová, Oldřich Navrátil, Jiban Kumar Kundu, and et al. 2022. "One-Enzyme RTX-PCR for the Detection of RNA Viruses from Multiple Virus Genera and Crop Plants" Viruses 14, no. 2: 298. https://doi.org/10.3390/v14020298
APA StyleHoffmeisterová, H., Kratochvílová, K., Čeřovská, N., Slavíková, L., Dušek, J., Muller, K., Fousek, J., Plchová, H., Navrátil, O., Kundu, J. K., & Moravec, T. (2022). One-Enzyme RTX-PCR for the Detection of RNA Viruses from Multiple Virus Genera and Crop Plants. Viruses, 14(2), 298. https://doi.org/10.3390/v14020298