
A Novel Coronavirus and a Broad Range of Viruses in Kenyan Cave Bats
 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
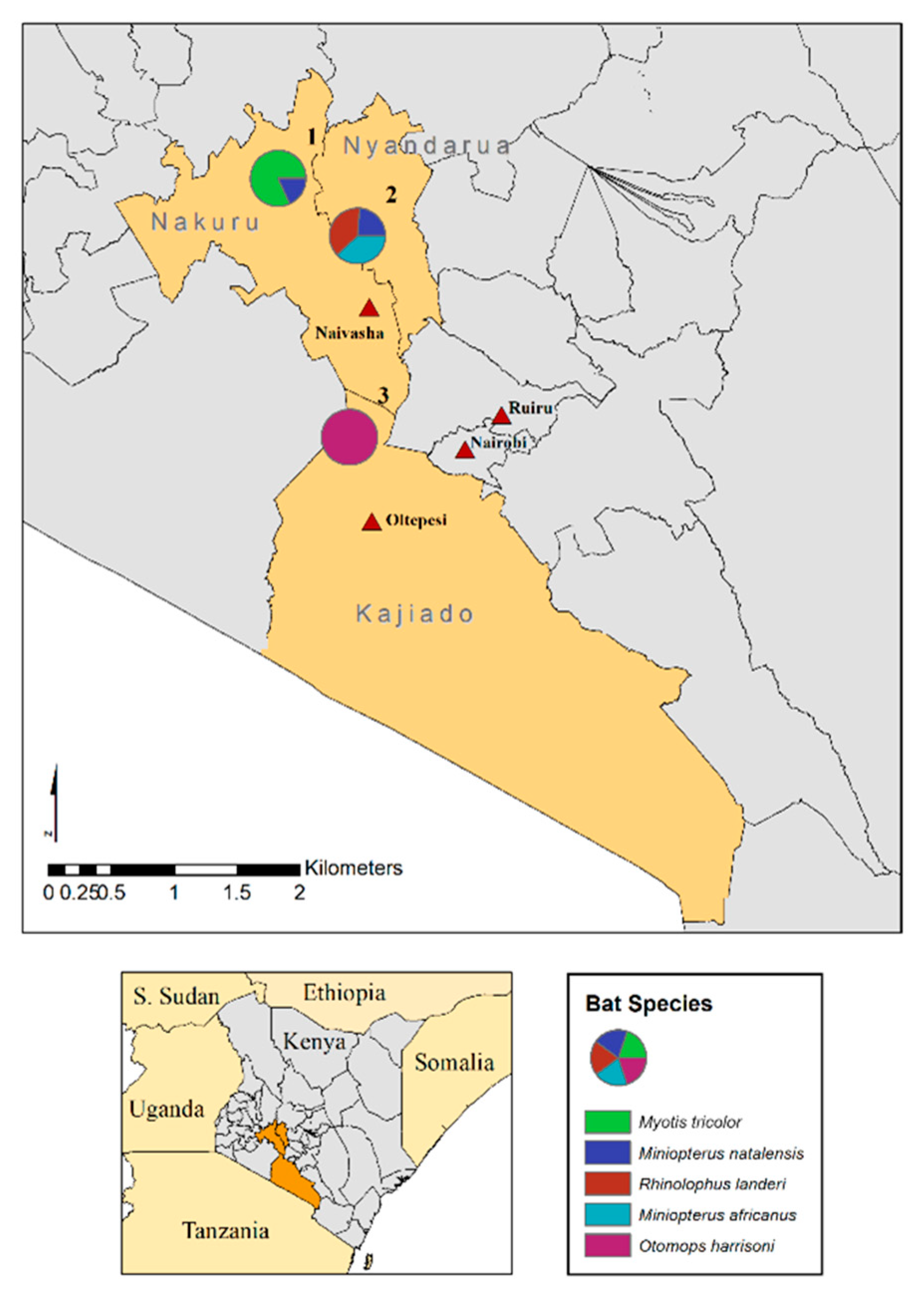
2.1. Bat Capture, Handling and Sampling
2.2. Sample Processing, Metagenome Sequencing and Data Analysis
3. Results
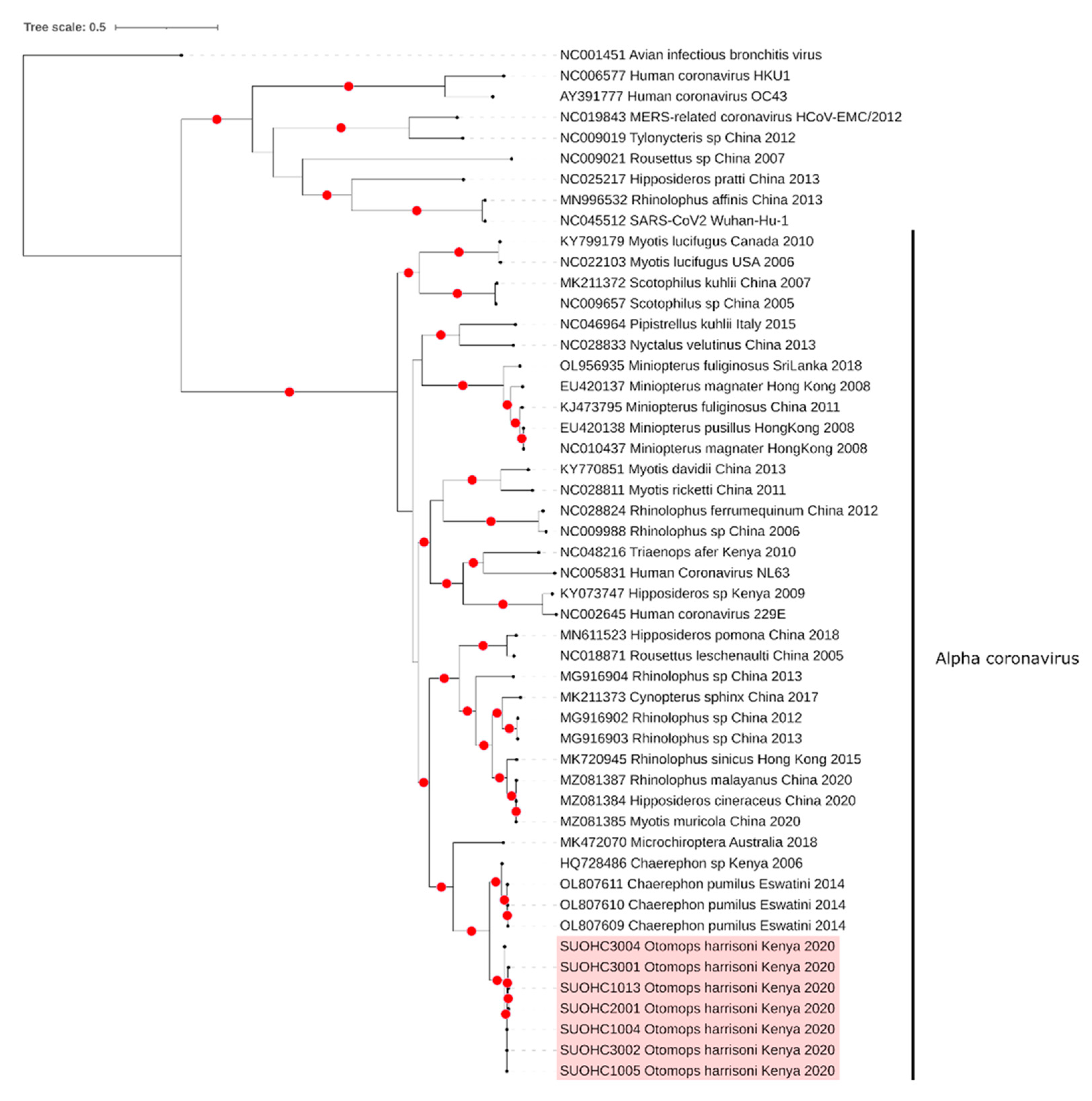
3.1. Coronaviruses
3.2. Retroviruses
3.3. Astrovirus and Other Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, L.H.; Latham, S.M.; Woolhouse, M.E.J. Risk factors for human disease emergence. Philos. Trans. R Soc. Lond. B Biol. Sci. 2001, 356, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.-C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef]
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many species of mammals are there? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef]
- Simmons, N.B.; Cirranello, A.L. Bat Species of the World: A Taxonomic and Geographic Database. 2022. Available online: https://batnames.org/ (accessed on 9 May 2022).
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Han, H.-J.; Wen, H.-L.; Zhou, C.-M.; Chen, F.-F.; Luo, L.-M.; Liu, J.-W.; Yu, X.-J. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Van Brussel, K.; Holmes, E.C. Zoonotic disease and virome diversity in bats. Curr. Opin. Virol. 2022, 52, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Nitsche, A.; Kurth, A. Update on Potentially Zoonotic Viruses of European Bats. Vaccines 2021, 9, 690. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Brinkmann, A.; Radonić, A.; Dabrowski, P.W.; Mühldorfer, K.; Nitsche, A.; Wibbelt, G.; Kurth, A. The virome of German bats: Comparing virus discovery approaches. Sci. Rep. 2021, 11, 7430. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.D.; Webala, P.W. Keys to the bats (Mammalia: Chiroptera) of East Africa. Fieldiana Life Earth Sci. 2012, 6, 1–60. [Google Scholar] [CrossRef]
- Musila, S.; Monadjem, A.; Webala, P.W.; Patterson, B.D.; Hutterer, R.; De Jong, Y.A.; Butynski, T.M.; Mwangi, G.; Chen, Z.-Z.; Jiang, X.-L. An annotated checklist of mammals of Kenya. Zool. Res. 2019, 40, 3–52. [Google Scholar] [CrossRef]
- Conrardy, C.; Tao, Y.; Kuzmin, I.V.; Niezgoda, M.; Agwanda, B.; Breiman, R.F.; Anderson, L.J.; Rupprecht, C.E.; Tong, S. Molecular detection of adenoviruses, rhabdoviruses, and paramyxoviruses in bats from Kenya. Am. J. Trop. Med. Hyg. 2014, 91, 258–266. [Google Scholar] [CrossRef]
- Kareinen, L.; Ogola, J.; Kivistö, I.; Smura, T.; Aaltonen, K.; Jääskeläinen, A.J.; Kibiwot, S.; Masika, M.M.; Nyaga, P.; Mwaengo, D.; et al. Range Expansion of Bombali Virus in Mops condylurus Bats, Kenya. Emerg. Infect. Dis. 2020, 26, 3007–3010. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Niezgoda, M.; Franka, R.; Agwanda, B.; Markotter, W.; Breiman, R.F.; Shieh, W.-J.; Zaki, S.R.; Rupprecht, C.E. Marburg virus in fruit bat, Kenya. Emerg. Infect. Dis. 2010, 16, 352–354. [Google Scholar] [CrossRef]
- Tao, Y.; Tang, K.; Shi, M.; Conrardy, C.; Li, K.S.; Lau, S.K.; Anderson, L.J.; Tong, S. Genomic characterization of seven distinct bat coronaviruses in Kenya. Virus Res. 2012, 167, 67–73. [Google Scholar] [CrossRef]
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482–485. [Google Scholar] [CrossRef]
- Waruhiu, C.; Ommeh, S.; Obanda, V.; Agwanda, B.; Gakuya, F.; Ge, X.Y.; Yang, X.L.; Wu, L.J.; Zohaib, A.; Hu, B.; et al. Molecular detection of viruses in Kenyan bats and discovery of novel astroviruses, caliciviruses and rotaviruses. Virol. Sin. 2017, 32, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.F.; King, R.; et al. IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wu, T.D.; Reeder, J.; Lawrence, M.; Beckerm, G.; Brauer, M.J. GMAP and GSNAP for Genomic Sequence Alignment: Enhancements to Speed, Accuracy, and Functionality. Methods Mol. Biol. 2016, 1418, 283–334. [Google Scholar]
- Altschul, S.F.; Gish, W.; Millerm, W.; Myers, E.W.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4678. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2, New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2, Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and Analyzing Genetic Recombination Using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar] [PubMed]
- Shapiro, J.T.; Mollerup, S.; Jensen, R.H.; Olofsson, J.K.; Nguyen, N.D.; Hansen, T.A.; Vinner, L.; Monadjem, A.; McCleery, R.A.; Hansen, A.J. Metagenomic analysis reveals previously undescribed bat coronavirus strains in Eswatini. Ecohealth 2021, 18, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.P.; Gao, Y.T.; Ge, X.Y.; Zhang, Q.; Peng, C.; Yang, X.L.; Tan, B.; Chen, J.; Chmura, A.A.; Daszak, P.; et al. Bat severe acute respiratory syndrome-like coronavirus WIV1 encodes an extra accessory protein, ORFX, involved in modulation of the host immune response. J. Virol. 2016, 90, 6573–6582. [Google Scholar] [CrossRef]
- Tristem, M.; Kabat, P.; Lieberman, L.; Linde, S.; Karpas, A.; Hill, F. Characterization of a novel murine leukemia virus-related subgroup within mammals. J. Virol. 1996, 70, 8241–8246. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef]
- Zhang, H.; Todd, S.; Tachedjian, M.; Barr, J.A.; Luo, M.; Yu, M.; Marsh, G.A.; Crameri, G.; Wang, L.-F. A novel bat herpesvirus encodes homologues of major histocompatibility complex classes I and II, C-type lectin, and a unique family of immune-related genes. J. Virol. 2012, 86, 8014–8030. [Google Scholar] [CrossRef]
- Leijonhufvud, G.; Bajalan, A.; Soratto, T.A.T.; Gustafsson, B.; Bogdanovic, G.; Bjerkner, A.; Allander, T.; Ljungman, G.; Andersson, B. Better detection of Torque teno virus in children with leukemia by metagenomic sequencing than by quantitative PCR. J. Med. Virol. 2022, 94, 634–641. [Google Scholar] [CrossRef]
- Kumar, A.; Murthy, S.; Kapoor, A. Evolution of selective-sequencing approaches for virus discovery and virome analysis. Virus Res. 2017, 239, 172–179. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Cui, J.; Tachedjian, M.; Wang, L.; Tachedjian, G.; Wang, L.F.; Zhang, S. Discovery of retroviral homologs in bats: Implications for the origin of mammalian gammaretroviruses. J. Virol. 2012, 86, 4288–4293. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Tachedjian, G.; Wang, L.F. Bats and rodents shape mammalian retroviral phylogeny. Sci. Rep. 2015, 5, 16561. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Tachedjian, G. Retroviruses of bats: A threat waiting in the wings? MBio 2021, 12, e01941-21. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, A.M.; Gifford, R.J. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PloS Biol. 2013, 11, e1001642. [Google Scholar] [CrossRef]
- De Benedictis, P.; Schultz-Cherry, S.; Burnham, A.; Cattoli, G. Astrovirus infections in humans and animals—Molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 2011, 11, 1529–1544. [Google Scholar] [CrossRef]
- Fischer, K.; Dos Reis, P.V.; Balkema-Buschmann, A. Bat Astroviruses: Towards Understanding the Transmission Dynamics of a Neglected Virus Family. Viruses 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Shioda, K.; Cosmas, L.; Audi, A.; Gregoricus, N.; Vinje, J.; Parashar, U.D.; Montgomery, J.M.; Feikin, D.R.; Breiman, R.F.; Hall, A.J. Population-Based Incidence Rates of Diarrheal Disease Associated with Norovirus, Sapovirus, and Astrovirus in Kenya. PLoS ONE 2016, 11, e0145943. [Google Scholar] [CrossRef]
- Amimo, J.O.; Machuka, E.M.; Abworo, E.O.; Vlasova, A.N.; Pelle, R. Whole Genome sequence analysis of porcine astroviruses reveals novel genetically diverse strains circulating in East African smallholder pig farms. Viruses 2020, 12, 1262. [Google Scholar] [CrossRef]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Sasaki, M.; Orba, Y.; Ueno, K.; Ishii, A.; Moonga, L.; Hang’Ombe, B.M.; Mweene, A.S.; Ito, K.; Sawa, H. Metagenomic analysis of the shrew enteric virome reveals novel viruses related to human stool-associated viruses. J. Gen. Virol. 2015, 96, 440–452. [Google Scholar] [CrossRef]
- Moon, J.S.; Domier, L.L.; McCoppin, N.K.; D’Arcy, C.J.; Jin, H. Nucleotide sequence analysis shows that Rhopalosiphum padi virus is a member of a novel group of in-sect-infecting RNA viruses. Virology 1998, 243, 54–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Y.; Hu, B.; Agwanda, B.; Fang, Y.; Wang, J.; Kuria, S.; Yang, J.; Masika, M.; Tang, S.; Lichoti, J.; et al. Viromes and surveys of RNA viruses in camel-derived ticks revealing transmission patterns of novel tick-borne viral pathogens in Kenya. Emerg. Microbes Infect. 2021, 10, 1975–1987. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, M.; Huang, Q.; Wang, Y.; Sun, J.; Zhang, Y.; Hou, C.; Wu, Y. Occurrence and distribution of Actinidia viruses in Shaanxi province of China. Plant Dis. 2021, 105, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Zana, B.; Kemenesi, G.; Urbán, P.; Földes, F.; Görföl, T.; Estók, P.; Boldogh, S.; Kurucz, K.; Jakab, F. Metagenomic analysis of bat guano samples revealed the presence of viruses potentially carried by insects, among others by Apis mellifera in Hungary. Acta Vet. Hung. 2018, 66, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Vasilakis, N.; Tian, J.-H.; Li, C.-X.; Chen, L.-J.; Eastwood, G.; Diao, X.-N.; Chen, M.-H.; Chen, X.; et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 2015, 90, 659–669. [Google Scholar] [CrossRef]
- Bennett, A.J.; Bushmaker, T.; Cameron, K.; Ondzie, A.; Niama, F.R.; Parra, H.-J.; Mombouli, J.-V.; Olson, S.H.; Munster, V.J.; Goldberg, T.L. Diverse RNA viruses of arthropod origin in the blood of fruit bats suggest a link between bat and arthropod viromes. Virology 2019, 528, 64–72. [Google Scholar] [CrossRef]
- James, S.; Donato, D.; de Thoisy, B.; Lavergnem, A.; Lacostem, V. Novel herpesviruses in neotropical bats and their relationship with other members of the Herpesviridae family. Infect Genet Evol 2020, 84, 104–367. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Viruses | ||||||||
|---|---|---|---|---|---|---|---|---|
| Site | Bat Species | Astroviridae (Bat Astrovirus) | Circoviridae (Cyclovirus) | Coronaviridae (Bat Alphacoronavirus) | Dicistroviridae (Rhopalosiphum Padi Virus) | Herpesviridae (Bat Herpesvirus) | Retroviridae1 | Unclassified 2 |
| Menengai Crater | Myotis tricolor (n = 9) | 1 | - | - | - | - | 9 | - |
| Miniopterus natalensis (n = 2) | 1 | - | - | 1 | - | - | 1 (Actinidia yellowing virus 2) | |
| Soysambu Conservancy | Miniopterus natalensis (n = 5) | 4 | 1 | - | - | - | 1 | - |
| Rhinolophus landeri (n = 8) | 2 | - | 1 | - | - | 8 | - | |
| Miniopterus africanus (n = 8) | 6 | - | - | - | 1 | 1 | 1 (Bole tick virus 4) | |
| Mount Suswa | Otomops harrisoni (n = 22) | - | - | 12 | - | - | 21 | - |
| Total | 54 | 14 (25.9%) | 1 (1.8%) | 13 (24.1%) | 1 (1.8%) | 1 (1.8%) | 40 (74.1%) | 2 (3.7%) |
| Retroviruses | ||||||||
|---|---|---|---|---|---|---|---|---|
| Site | Bat Species | Myotis Myotis Endogenous Retrovirus (MMER) | Myotis Myotis Endogenous Retrovirus (MDER) | Myotis Myotis Endogenous Retrovirus (MRER) | Rhinolophus Ferrumequinum Retrovirus (RFRV) | Galidia Endogenous Retrovirus (GERV) | Murine leukemia Related Virus (MLRV) | Rhinolophus Affinis Foamy Virus 1 (RAFV1) |
| Menengai Crater | Myotis tricolor (n = 9) | 9 | 1 | 1 | - | - | - | - |
| Miniopterus natalensis (n = 2) | - | - | - | - | - | - | - | |
| Soysambu Conservancy | Miniopterus natalensis (n = 5) | - | - | - | 1 | - | - | - |
| Rhinolophus landeri (n = 8) | - | - | - | 8 | - | - | - | |
| Miniopterus africanus (n = 8) | - | - | - | 1 | - | - | - | |
| Mount Suswa | Otomops harrisoni (n = 22) | - | - | - | - | 19 | 16 | 1 |
| Total | 54 | 9 (16.6%) | 1 (1.8%) | 1 (1.8%) | 10 (18.5%) | 19 (35.1%) | 16 (29.6%) | 1 (1.8%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamau, J.; Ergunay, K.; Webala, P.W.; Justi, S.A.; Bourke, B.P.; Kamau, M.W.; Hassell, J.; Chege, M.N.; Mwaura, D.K.; Simiyu, C.; et al. A Novel Coronavirus and a Broad Range of Viruses in Kenyan Cave Bats. Viruses 2022, 14, 2820. https://doi.org/10.3390/v14122820
Kamau J, Ergunay K, Webala PW, Justi SA, Bourke BP, Kamau MW, Hassell J, Chege MN, Mwaura DK, Simiyu C, et al. A Novel Coronavirus and a Broad Range of Viruses in Kenyan Cave Bats. Viruses. 2022; 14(12):2820. https://doi.org/10.3390/v14122820
Chicago/Turabian StyleKamau, Joseph, Koray Ergunay, Paul W. Webala, Silvia A. Justi, Brian P. Bourke, Maureen W. Kamau, James Hassell, Mary N. Chege, David K. Mwaura, Cynthia Simiyu, and et al. 2022. "A Novel Coronavirus and a Broad Range of Viruses in Kenyan Cave Bats" Viruses 14, no. 12: 2820. https://doi.org/10.3390/v14122820
APA StyleKamau, J., Ergunay, K., Webala, P. W., Justi, S. A., Bourke, B. P., Kamau, M. W., Hassell, J., Chege, M. N., Mwaura, D. K., Simiyu, C., Kibiwot, S., Onyuok, S., Caicedo-Quiroga, L., Li, T., Zimmerman, D. M., & Linton, Y.-M. (2022). A Novel Coronavirus and a Broad Range of Viruses in Kenyan Cave Bats. Viruses, 14(12), 2820. https://doi.org/10.3390/v14122820