
Development and Visualization Improvement for the Rapid Detection of Decapod Iridescent Virus 1 (DIV1) in Penaeus vannamei Based on an Isothermal Recombinase Polymerase Amplification Assay

Abstract
1. Introduction
2. Materials and Methods
2.1. Pathogen Samples and Recombinant Plasmid Construction
2.2. Primer and Probe Design
2.3. DIV1-qPCR and DIV1-qRPA Assay
2.4. DIV1-RPA-SYBR Green I Assay
2.5. Evaluation of Sensitivity and Specificity
3. Results
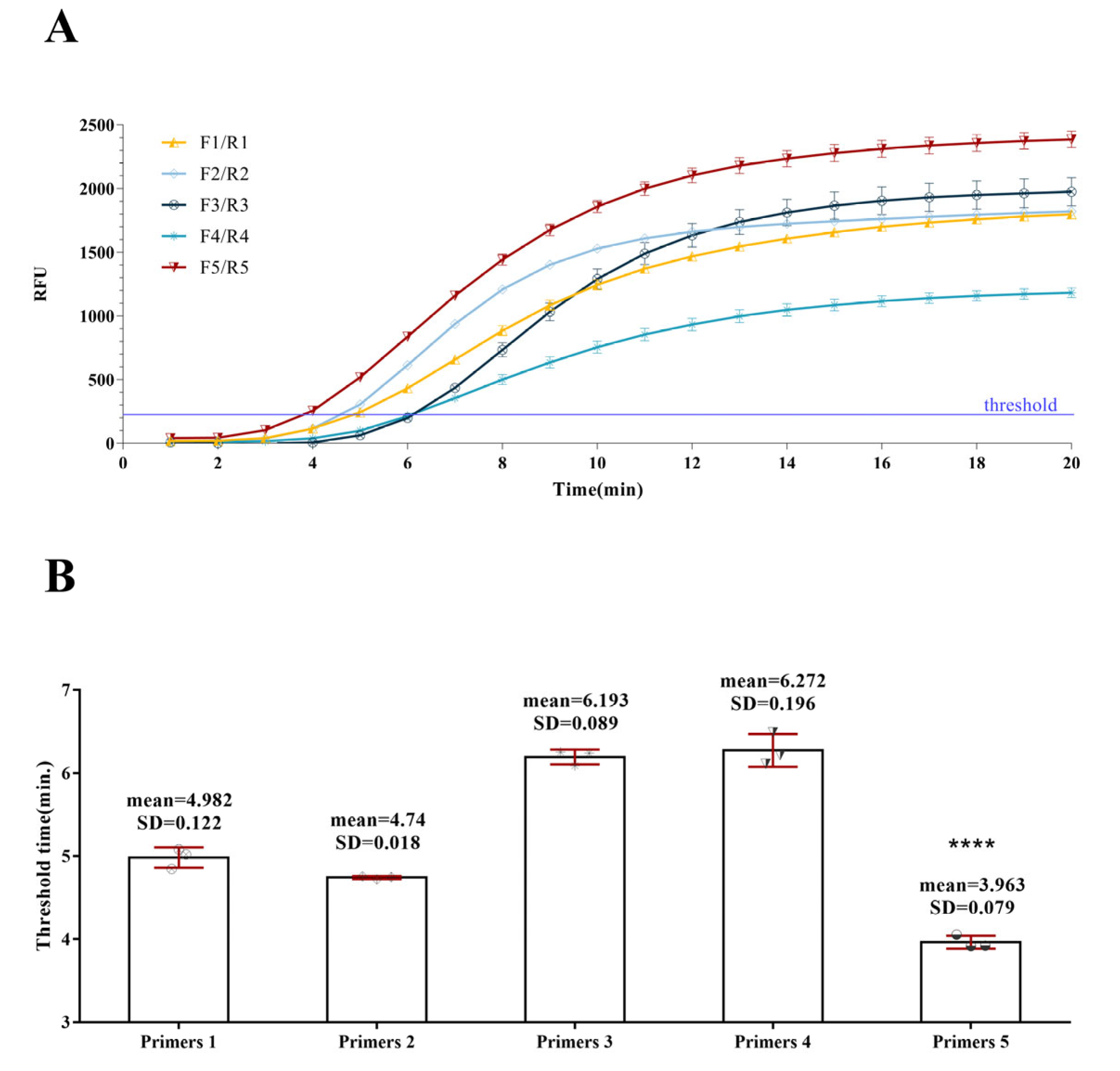
3.1. Primer Screening
3.2. Optimizing the Reaction Temperature of DIV1-qRPA Assay
3.3. Optimizing the Probe Consumption of the DIV1-qRPA Assay
3.4. Sensitivity Evaluation of qPCR and qRPA Assays
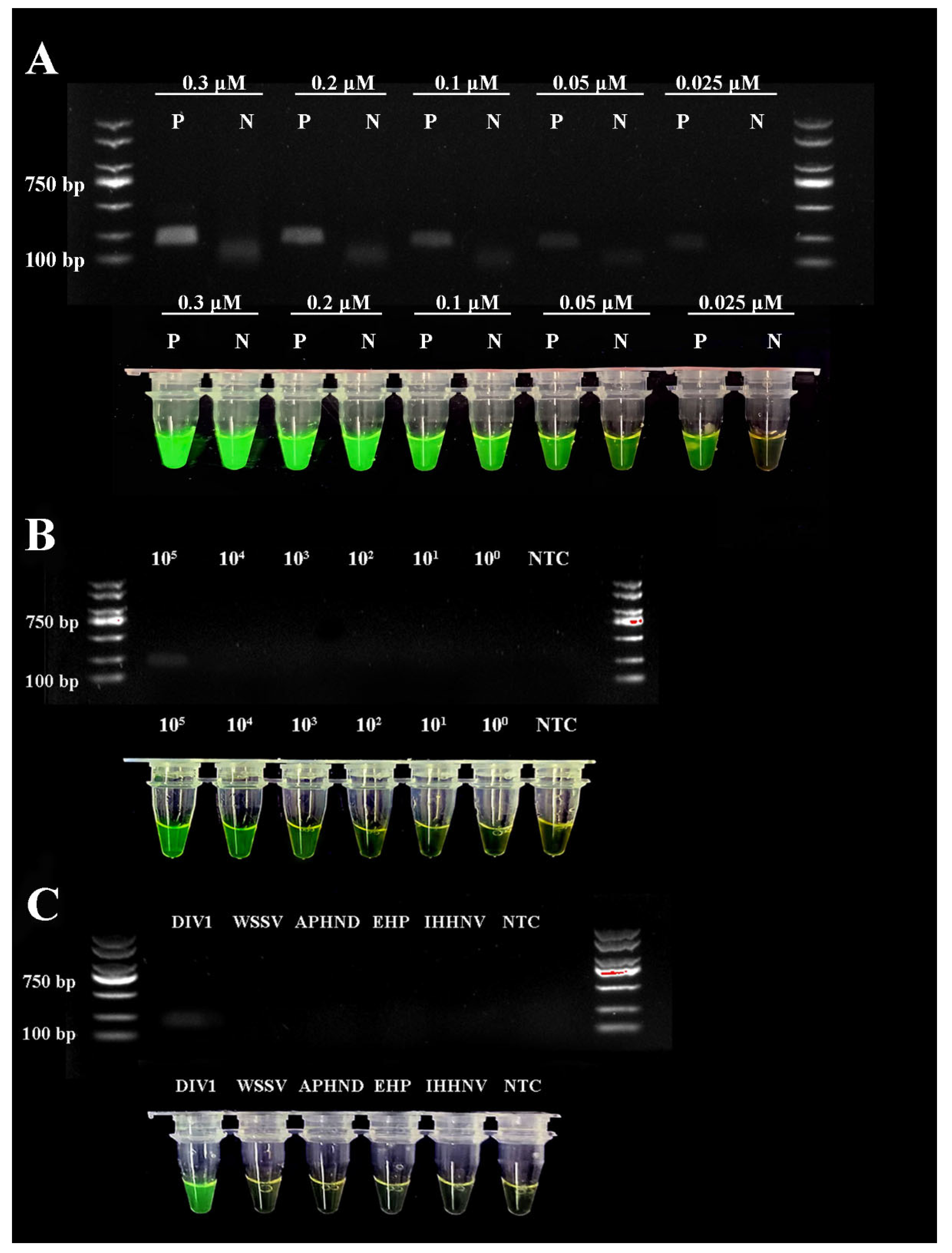
3.5. Optimizing the Primer Concentrations of DIV1-RPA-SYBR Green I Assay
3.6. Sensitivity Evaluation of RPA-SYBR Green I Assay
3.7. Specificity Evaluation of qRPA and RPA-SYBR Green I Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharawy, Z.Z.; Ashour, M.; Labena, A.; Alsaqufi, A.S.; Mansour, A.T.; Abbas, E.M. Effects of Dietary Arthrospira platensis Nanoparticles on Growth Performance, Feed Utilization, and Growth-Related Gene Expression of Pacific White Shrimp, Litopenaeus vannamei. Aquaculture 2022, 551, 737905. [Google Scholar] [CrossRef]
- Mansour, A.T.; Ashour, M.; Abbas, E.M.; Alsaqufi, A.S.; Kelany, M.S.; El-Sawy, M.A.; Sharawy, Z.Z. Growth Performance, Immune-Related and Antioxidant Genes Expression, and Gut Bacterial Abundance of Pacific White Leg Shrimp, Litopenaeus vannamei, Dietary Supplemented with Natural Astaxanthin. Front. Physiol. 2022, 13, 1245. [Google Scholar] [CrossRef]
- FAO. The State of World Fisheries and Aquaculture 2020; Food and Agriculture Organization of the United Nations: Rome, Italy, 2020. [Google Scholar] [CrossRef]
- Xu, L.; Wang, T.; Li, F.; Yang, F. Isolation and Preliminary Characterization of a New Pathogenic Iridovirus from Redclaw Crayfish Cherax Quadricarinatus. Dis. Aquat. Org. 2016, 120, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Chen, M.M.; Wan, X.Y.; Li, C.; Zhang, Q.L.; Wang, R.Y.; Cheng, D.Y.; Dong, X.; Yang, B.; Wang, X.H.; et al. Characterization of a New Member of Iridoviridae, Shrimp Hemocyte Iridescent Virus (SHIV), Found in White Leg Shrimp (Litopenaeus vannamei). Sci. Rep. 2017, 7, 11834. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Hyatt, A.; Miyazaki, T.; Williams, T. Family Iridoviridae: Poor Viral Relations No Longer. Curr. Top. Microbiol. Immunol. 2009, 328, 123–170. [Google Scholar] [CrossRef]
- NACA Network of Aquaculture Centres in Asia-Pacific. Diseases of Crustaceans–Infection with Decapod Iridescent Virus 1 (DIV1). 2020. Available online: https://www.researchgate.net/publication/341600253_Disease_Advisory_Decapod_iridescent_virus_1_DIV1_an_emerging_threat_to_the_shrimp_industry (accessed on 18 June 2021).
- Chen, X.; Qiu, L.; Wang, H.; Zou, P.; Dong, X.; Li, F.; Huang, J. Susceptibility of Exopalaemon Carinicauda to the Infection with Shrimp Hemocyte Iridescent Virus (SHIV 20141215), a Strain of Decapod Iridescent Virus 1 (DIV1). Viruses 2019, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.K.; Yuan, H.F.; Wang, T.T.; Yang, F.; Chen, J.M. Study of Cherax Quadricarinatus Iridescent Virus in Two Crabs. J. Appl. Oceanogr. 2017, 1, 82–86. [Google Scholar]
- Qiu, L.; Chen, X.; Zhao, R.H.; Li, C.; Gao, W.; Zhang, Q.L.; Huang, J. Description of a Natural Infection with Decapod Iridescent Virus 1 in Farmed Giant Freshwater Prawn, Macrobrachium rosenbergii. Viruses 2019, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Srisala, J.; Sanguanrut, P.; Thaiue, D.; Laiphrom, S.; Siriwattano, J.; Khudet, J.; Powtongsook, S.; Flegel, T.W.; Sritunyalucksana, K. Infectious Myonecrosis Virus (IMNV) and Decapod Iridescent Virus 1 (DIV1) Detected in Captured, Wild Penaeus monodon. Aquaculture 2021, 545, 737262. [Google Scholar] [CrossRef]
- Qiu, L.; Chen, X.; Guo, X.M.; Gao, W.; Zhao, R.H.; Zhang, Q.L.; Yang, B.; Huang, J. A TaqMan Probe Based Real-Time PCR for the Detection of Decapod Iridescent Virus 1. J. Invertebr. Pathol. 2020, 173, 107367. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Chen, M.M.; Wan, X.Y.; Zhang, Q.L.; Li, C.; Dong, X.; Yang, B.; Huang, J. Detection and Quantification of Shrimp Hemocyte Iridescent Virus by TaqMan Probe Based Real-Time PCR. J. Invertebr. Pathol. 2018, 154, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.-y.; Li, Q.-y.; Zhang, H.; Ye, L.; Shi, L.; Feng, Y. hui Development and Comparison of QPCR and QLAMP for Rapid Detection of the Decapod Iridescent Virus 1 (DIV1). J. Invertebr. Pathol. 2021, 182, 107567. [Google Scholar] [CrossRef]
- Qiu, L.; Chen, M.M.; Wang, R.Y.; Wan, X.Y.; Li, C.; Zhang, Q.L.; Dong, X.; Yang, B.; Xiang, J.H.; Huang, J. Complete Genome Sequence of Shrimp Hemocyte Iridescent Virus (SHIV) Isolated from White Leg Shrimp, Litopenaeus vannamei. Arch. Virol. 2018, 163, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Abd El Wahed, A.; El-Deeb, A.; El-Tholoth, M.; Abd El Kader, H.; Ahmed, A.; Hassan, S.; Hoffmann, B.; Haas, B.; Shalaby, M.A.; Hufert, F.T.; et al. A Portable Reverse Transcription Recombinase Polymerase Amplification Assay for Rapid Detection of Foot-and-Mouth Disease Virus. PLoS ONE 2013, 8, e71642. [Google Scholar] [CrossRef]
- Amer, H.M.; Abd El Wahed, A.; Shalaby, M.A.; Almajhdi, F.N.; Hufert, F.T.; Weidmann, M. A New Approach for Diagnosis of Bovine Coronavirus Using a Reverse Transcription Recombinase Polymerase Amplification Assay. J. Virol. Methods 2013, 193, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Crannell, Z.A.; Rohrman, B.; Richards-Kortum, R. Equipment-Free Incubation of Recombinase Polymerase Amplification Reactions Using Body Heat. PLoS ONE 2014, 9, e112146. [Google Scholar] [CrossRef]
- Crannell, Z.A.; Cabada, M.M.; Castellanos-Gonzalez, A.; Irani, A.; White, A.C.; Richards-Kortum, R. Recombinase Polymerase Amplification-Based Assay to Diagnose Giardia in Stool Samples. Am. J. Trop. Med. Hyg. 2015, 92, 583–587. [Google Scholar] [CrossRef]
- Euler, M.; Wang, Y.; Nentwich, O.; Piepenburg, O.; Hufert, F.T.; Weidmann, M. Recombinase Polymerase Amplification Assay for Rapid Detection of Rift Valley Fever Virus. J. Clin. Virol. 2012, 54, 308–312. [Google Scholar] [CrossRef]
- Loo, J.F.C.; Lau, P.M.; Ho, H.P.; Kong, S.K. An Aptamer-Based Bio-Barcode Assay with Isothermal Recombinase Polymerase Amplification for Cytochrome-c Detection and Anti-Cancer Drug Screening. Talanta 2013, 115, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, L.; Jin, W.; Wan, Y. Recombinase Polymerase Amplification (RPA) of CaMV-35S Promoter and Nos Terminator for Rapid Detection of Genetically Modified Crops. Int. J. Mol. Sci. 2014, 15, 18197–18205. [Google Scholar] [CrossRef] [PubMed]
- Babu, B.; Ochoa-Corona, F.M.; Paret, M.L. Recombinase Polymerase Amplification Applied to Plant Virus Detection and Potential Implications. Anal. Biochem. 2018, 546, 72–77. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase Polymerase Amplification: Basics, Applications and Recent Advances. TrAC-Trends Anal. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Wambua, L.; Schneider, B.; Okwaro, A.; Wanga, J.O.; Imali, O.; Wambua, P.N.; Agutu, L.; Olds, C.; Jones, C.S.; Masiga, D.; et al. Development of Field-Applicable Tests for Rapid and Sensitive Detection of Candidatus Phytoplasma Oryzae. Mol. Cell. Probes 2017, 35, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, P.J.; Suebsing, R.; Sirithammajak, S.; Kampeera, J.; Turner, W.; Jeffs, A.; Kiatpathomchai, W.; Withyachumanarnkul, B. Vertical Transmission and Concurrent Infection of Multiple Bacterial Pathogens in Naturally Infected Red Tilapia (Oreochromis Spp.). Aquac. Res. 2017, 48, 2706–2717. [Google Scholar] [CrossRef]
- Mabrok, M.; Elayaraja, S.; Chokmangmeepisarn, P.; Jaroenram, W.; Arunrut, N.; Kiatpathomchai, W.; Debnath, P.P.; Delamare-Deboutteville, J.; Mohan, C.V.; Fawzy, A.; et al. Rapid Visualization in the Specific Detection of Flavobacterium Columnare, a Causative Agent of Freshwater Columnaris Using a Novel Recombinase Polymerase Amplification (RPA) Combined with Lateral Flow Dipstick (LFD) Assay. Aquaculture 2021, 531, 735780. [Google Scholar] [CrossRef]
- Wang, H.; Dong, J.; Zhang, T.; Wang, F.; Yang, R.; Zhang, Y.; Zhao, X. A Novel Rapid Detection of Senecavirus A Using Recombinase Polymerase Amplification (RPA) Coupled with Lateral Flow (LF) Dipstrip. Anal. Biochem. 2022, 646, 114627. [Google Scholar] [CrossRef]
- Liu, Y.; Lei, T.; Liu, Z.; Kuang, Y.; Lyu, J.; Wang, Q. A Novel Technique to Detect EGFR Mutations in Lung Cancer. Int. J. Mol. Sci. 2016, 17, 792. [Google Scholar] [CrossRef]
- Zheng, Y.; Hu, P.; Ren, H.; Wang, H.; Cao, Q.; Zhao, Q.; Li, H.; Zhang, H.; Liu, Z.; Li, Y.; et al. RPA-SYBR Green I Based Instrument-Free Visual Detection for Pathogenic Yersinia Enterocolitica in Meat. Anal. Biochem. 2021, 621, 114157. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, J.; Zhang, F.; Zhou, Y.; Huang, H. Detection of Shrimp Hemocyte Iridescent Virus by Recombinase Polymerase Amplification Assay. Mol. Cell. Probes 2020, 49, 101475. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Primer/Probe | Sequence (5′-3′) | Product Size (bp) |
|---|---|---|---|
| qPCR | FP | AGGAAAGGAAACGAAAGAAATTATACC | 87 |
| RP | GCTTGATCGGCATCCTTGA | ||
| PP | FAM-CACATGATTTGCAACAAGCTTCCAGCA-BHQ1 | ||
| qRPA | FR1 | GGTATCTTATTCAGAGATGGAAGACTATCAGG | 137 |
| RR1 | CTCTTGATGGATACACTGATCTTCGAATTG | ||
| FR2 | GTGAAATTGTAGAGCCAGAGATTGTAACGGTAG | 190 | |
| RR2 | CTGATTGGGATGGTCATATTAGGATACTCTTC | ||
| FR3 | GGAAGACTATCAGGAAGTGTGAAATTGTAGAG | 117 | |
| RR3 | CTTGATGGATACACTGATCTTCGAATTGCTTG | ||
| FR4 | GAAATTGTAGAGCCAGAGATTGTAACGGTAG | 182 | |
| RR4 | GGGATGGTCATATTAGGATACTCTTCTTTG | ||
| FR5 | GTGTGAAATTGTAGAGCCAGAGATTGTAAC | 190 | |
| RR5 | GATTGGGATGGTCATATTAGGATACTCTTC | ||
| PR | TAGAGCCAGAGATTGTAACGGTAGCTACATCT[dT-FAM]C[THF][dT-BHQ1]ACCGAAACGAAAACG(C3 spacer) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wang, Y.; Hu, J.; Bao, Z.; Wang, M. Development and Visualization Improvement for the Rapid Detection of Decapod Iridescent Virus 1 (DIV1) in Penaeus vannamei Based on an Isothermal Recombinase Polymerase Amplification Assay. Viruses 2022, 14, 2752. https://doi.org/10.3390/v14122752
Xu Y, Wang Y, Hu J, Bao Z, Wang M. Development and Visualization Improvement for the Rapid Detection of Decapod Iridescent Virus 1 (DIV1) in Penaeus vannamei Based on an Isothermal Recombinase Polymerase Amplification Assay. Viruses. 2022; 14(12):2752. https://doi.org/10.3390/v14122752
Chicago/Turabian StyleXu, Yajin, Yan Wang, Jingjie Hu, Zhenmin Bao, and Mengqiang Wang. 2022. "Development and Visualization Improvement for the Rapid Detection of Decapod Iridescent Virus 1 (DIV1) in Penaeus vannamei Based on an Isothermal Recombinase Polymerase Amplification Assay" Viruses 14, no. 12: 2752. https://doi.org/10.3390/v14122752
APA StyleXu, Y., Wang, Y., Hu, J., Bao, Z., & Wang, M. (2022). Development and Visualization Improvement for the Rapid Detection of Decapod Iridescent Virus 1 (DIV1) in Penaeus vannamei Based on an Isothermal Recombinase Polymerase Amplification Assay. Viruses, 14(12), 2752. https://doi.org/10.3390/v14122752