

Cell-Type-Dependent Role for nsP3 Macrodomain ADP-Ribose Binding and Hydrolase Activity during Chikungunya Virus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viruses, Infections, and Assays
2.3. Infectious Center Assays
2.4. Flow Cytometry for dsRNA
2.5. qRT-PCR for Viral RNA
2.6. Immunoblot Analyses of Viral Protein Expression
2.7. Pulse-chase Analysis of PE2 Protein Processing
2.8. Extracellular Genome: PFU Ratio
2.9. Puromycin Translation Assay
2.10. Statistical Analysis
3. Results
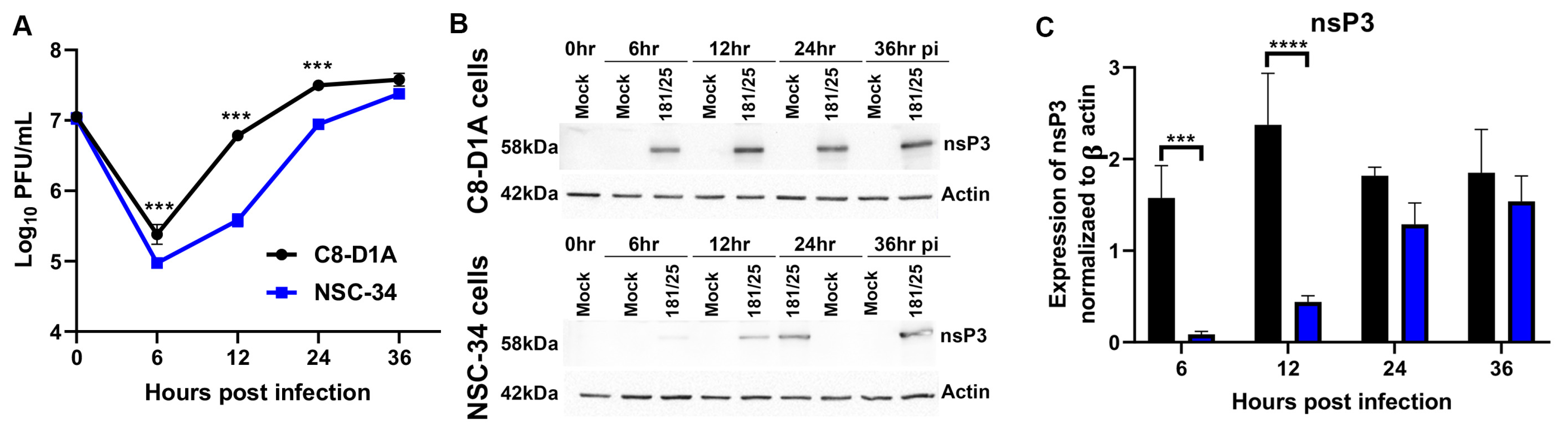
3.1. CHIKV Replicates more Efficiently in C8-D1A Cells than NSC-34 Cells
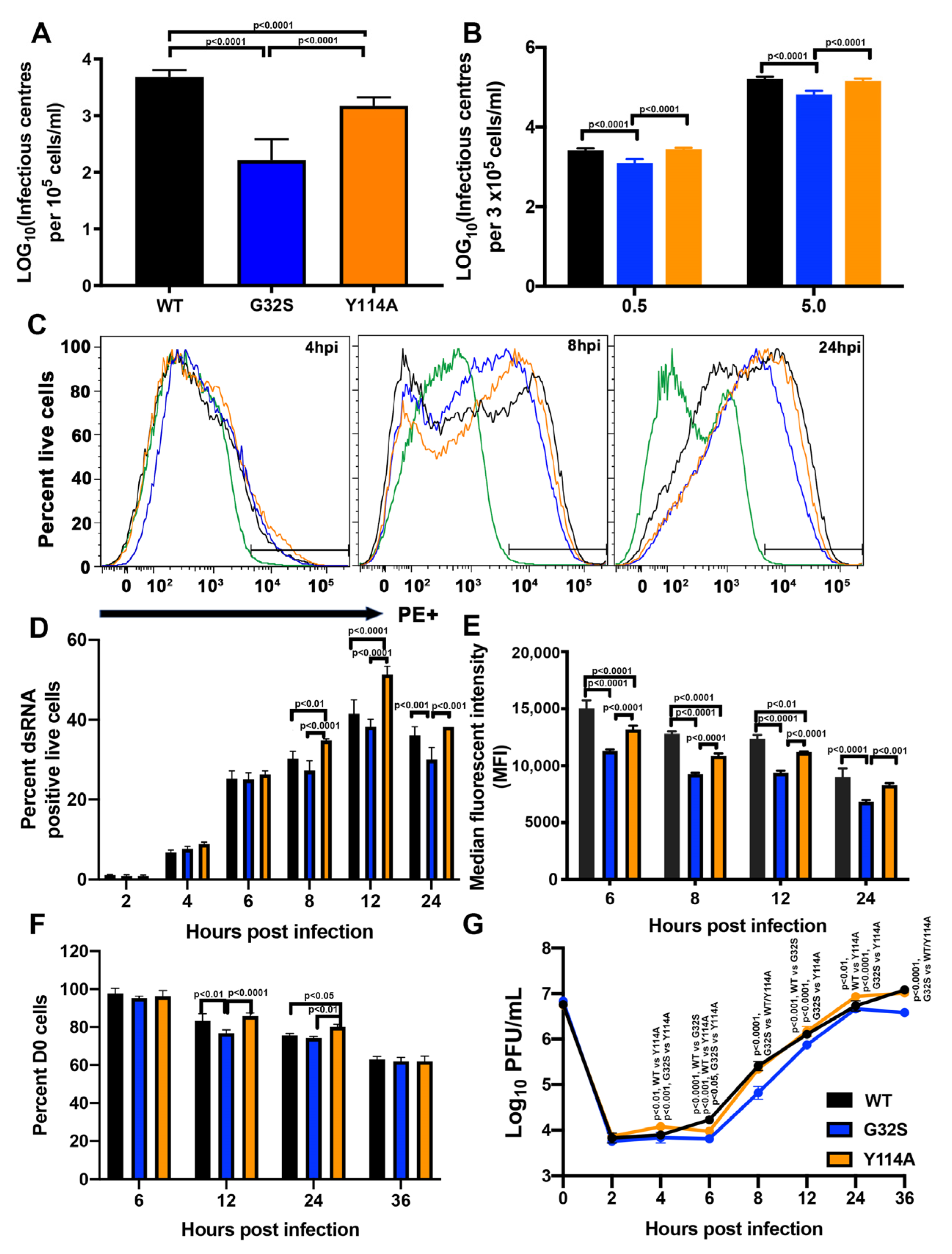
3.2. nsP3 MD Function Affects the Initiation of CHIKV Infection in C8-D1A Cells
3.3. nsP3 MD Function Affects Amplification of Replication Complexes and Virus Production by C8-D1A Cells
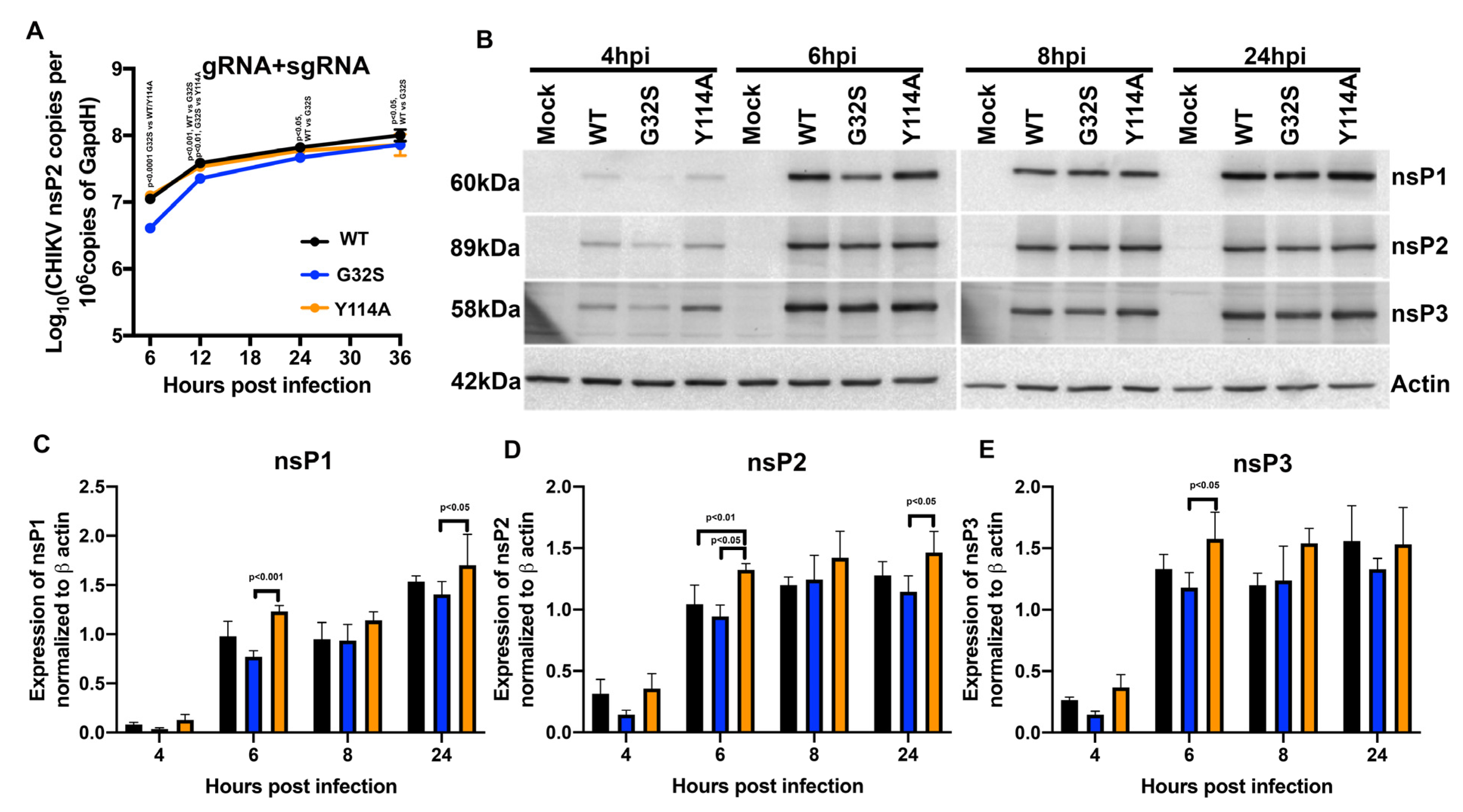
3.4. nsP3 MD ADPr Binding Affects gRNA Translation and Viral RNA Synthesis in C8-D1A Cells
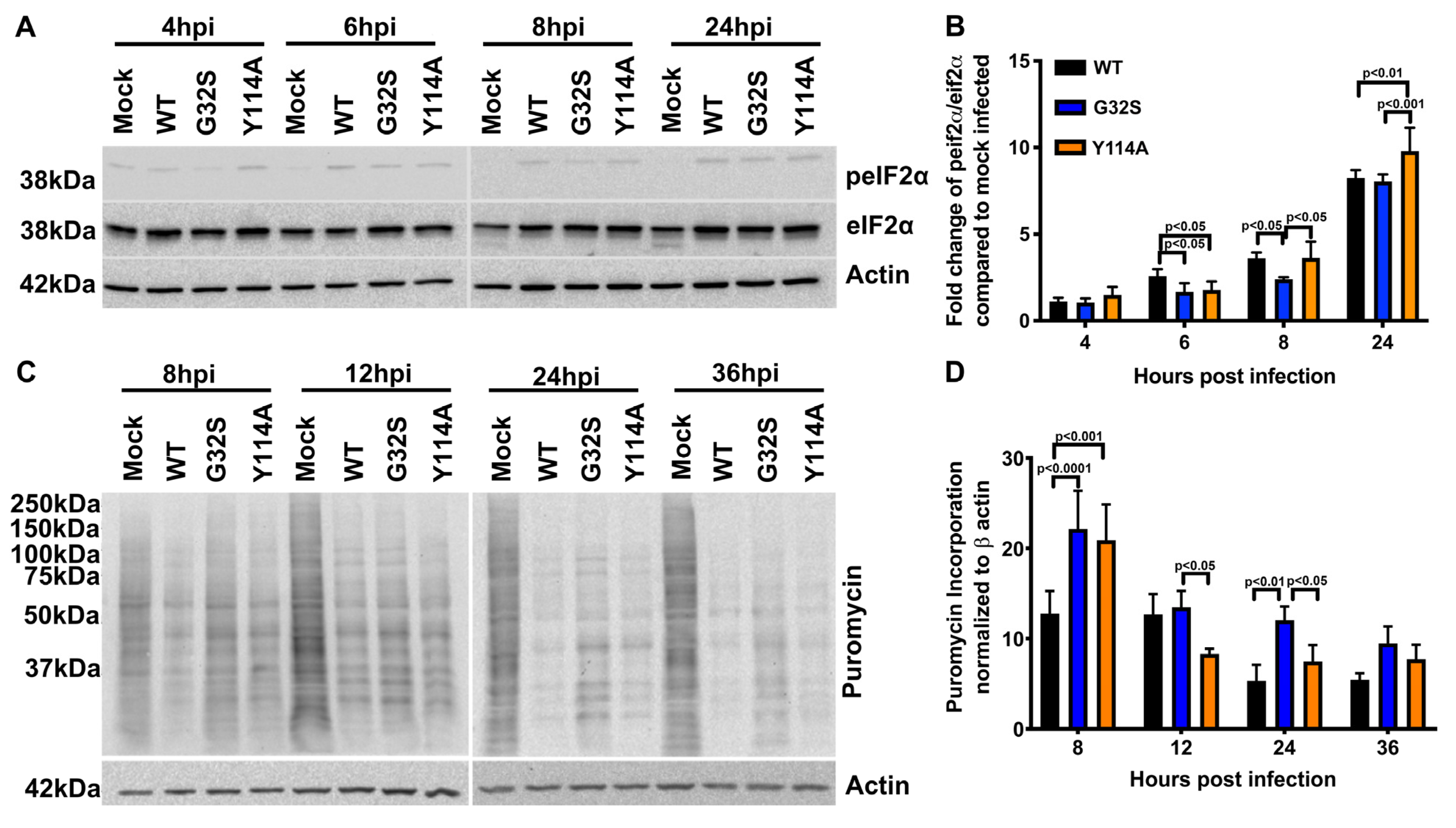
3.5. Effect of nsP3 MD Function on Host Translational Shut-off in CHIKV-infected C8-D1A Cells
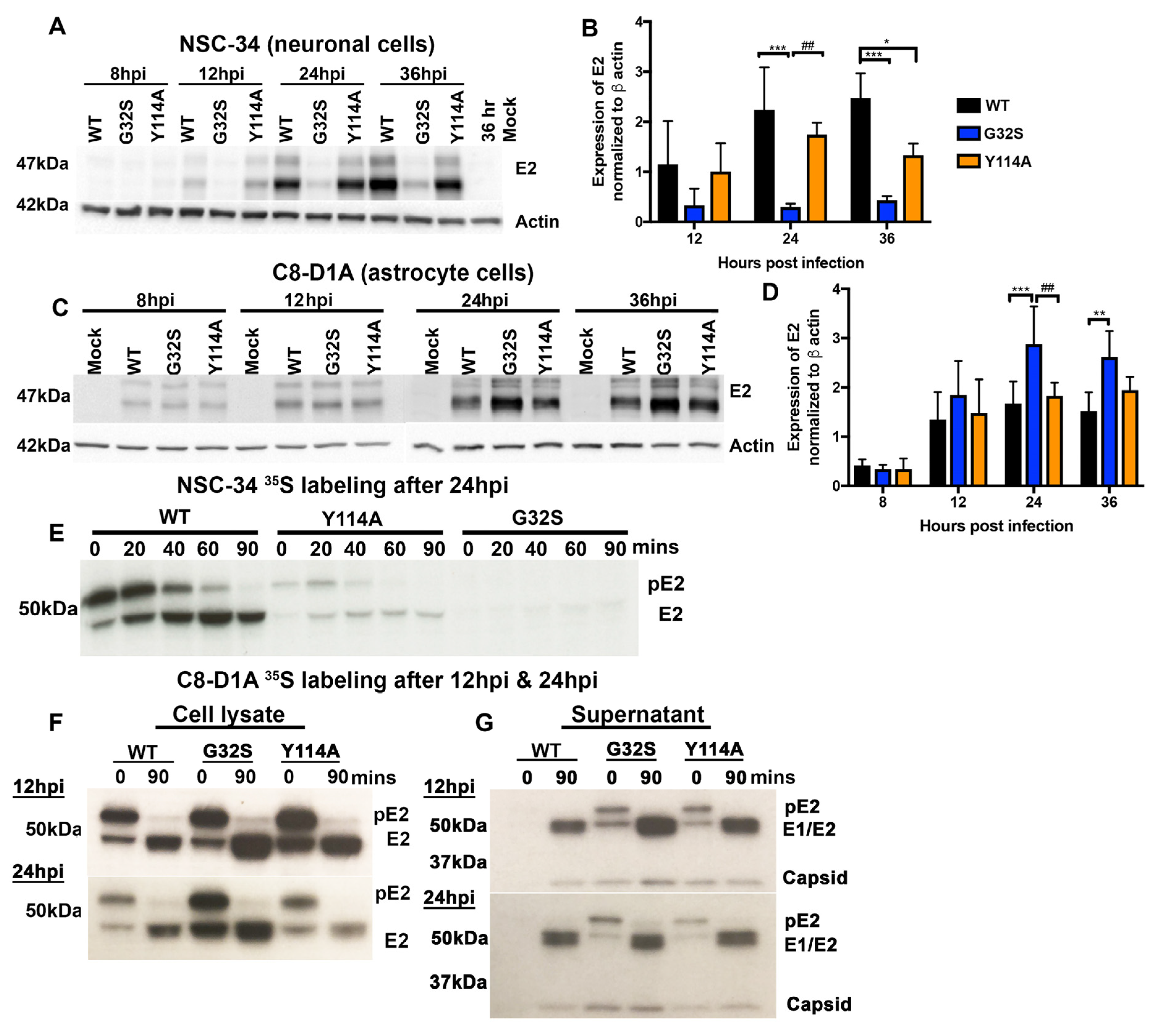
3.6. Translation of Structural Proteins from sgRNA in C8-D1A and NSC-34 Cells Is Differentially Affected by Altered nsP3 MD Function
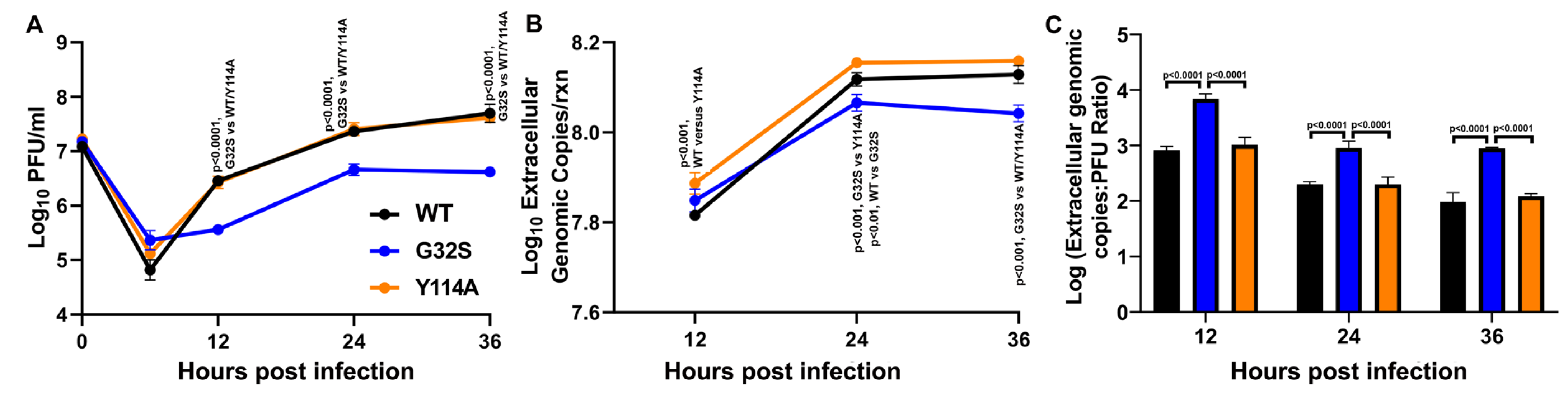
3.7. G32S Virions Produced by C8-D1A Cells Are less Infectious than WT or Y114A Virions
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Gerardin, P.; de Brito, C.A.A.; Soares, C.N.; Ferreira, M.L.B.; Solomon, T. The neurological complications of chikungunya virus: A systematic review. Rev. Med. Virol. 2018, 28, e1978. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Hoarau, J.J.; Jaffar Bandjee, M.C.; Maquart, M.; Gasque, P. Multifaceted innate immune responses engaged by astrocytes, microglia and resident dendritic cells against Chikungunya neuroinfection. J. Gen. Virol. 2015, 96, 294–310. [Google Scholar] [CrossRef] [PubMed]
- Inglis, F.M.; Lee, K.M.; Chiu, K.B.; Purcell, O.M.; Didier, P.J.; Russell-Lodrigue, K.; Weaver, S.C.; Roy, C.J.; MacLean, A.G. Neuropathogenesis of chikungunya infection: Astrogliosis and innate immune activation. J. Neurovirol. 2016, 22, 140–148. [Google Scholar] [CrossRef]
- Hucke, F.I.L.; Bestehorn-Willmann, M.; Bassetto, M.; Brancale, A.; Zanetta, P.; Bugert, J.J. CHIKV strains Brazil (wt) and Ross (lab-adapted) differ with regard to cell host range and antiviral sensitivity and show CPE in human glioblastoma cell lines U138 and U251. Virus Genes 2022, 58, 188–202. [Google Scholar] [CrossRef]
- Abraham, R.; Mudaliar, P.; Padmanabhan, A.; Sreekumar, E. Induction of cytopathogenicity in human glioblastoma cells by chikungunya virus. PLoS ONE 2013, 8, e75854. [Google Scholar] [CrossRef]
- Abraham, R.; Singh, S.; Nair, S.R.; Hulyalkar, N.V.; Surendran, A.; Jaleel, A.; Sreekumar, E. Nucleophosmin (NPM1)/B23 in the Proteome of Human Astrocytic Cells Restricts Chikungunya Virus Replication. J. Proteome Res. 2017, 16, 4144–4155. [Google Scholar] [CrossRef]
- Eleftheriadou, I.; Dieringer, M.; Poh, X.Y.; Sanchez-Garrido, J.; Gao, Y.; Sgourou, A.; Simmons, L.E.; Mazarakis, N.D. Selective transduction of astrocytic and neuronal CNS subpopulations by lentiviral vectors pseudotyped with chikungunya virus envelope. Biomaterials 2017, 123, 1–14. [Google Scholar] [CrossRef]
- Antel, J.P.; Becher, B.; Ludwin, S.K.; Prat, A.; Quintana, F.J. Glial cells as regulators of neuroimmune interactions in the central nervous system. J. Immunol. 2020, 204, 251–255. [Google Scholar] [CrossRef]
- Pfefferkorn, C.; Kallfass, C.; Lienenklaus, S.; Spanier, J.; Kalinke, U.; Rieder, M.; Conzelmann, K.K.; Michiels, T.; Staeheli, P. Abortively infected astrocytes appear to represent the main source of interferon beta in the virus-infected brain. J. Virol. 2015, 90, 2031–2038. [Google Scholar] [CrossRef]
- Bohmwald, K.; Andrade, C.A.; Galvez, N.M.S.; Mora, V.P.; Munoz, J.T.; Kalergis, A.M. The causes and long-term consequences of viral encephalitis. Front. Cell. Neurosci. 2021, 15, 755875. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.A.; Misasi, J.; Smole, S.; Feldman, H.A.; Cohen, A.B.; Santagata, S.; McManus, M.; Ahmed, A.A. Eastern equine encephalitis in children, Massachusetts and New Hampshire, USA, 1970–2010. Emerg. Infect. Dis. 2013, 19, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Garen, P.D.; Tsai, T.F.; Powers, J.M. Human eastern equine encephalitis: Immunohistochemistry and ultrastructure. Mod. Pathol. 1999, 12, 646–652. [Google Scholar] [PubMed]
- German, A.C.; Myint, K.S.; Mai, N.T.; Pomeroy, I.; Phu, N.H.; Tzartos, J.; Winter, P.; Collett, J.; Farrar, J.; Barrett, A.; et al. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model. Trans. Roy. Soc. Trop. Med. Hyg. 2006, 100, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Preusser, M.; Garzuly, F.; Holzmann, H.; Heinz, F.X.; Budka, H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J. Neuropathol. Exp. Neurol. 2005, 64, 506–512. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- LaPointe, A.T.; Sokoloski, K.J. De-coding the contributions of the viral RNAs to alphaviral pathogenesis. Pathogens 2021, 10, 771. [Google Scholar] [CrossRef]
- Hardy, W.R.; Strauss, J.H. Processing the nonstructural polyproteins of Sindbis virus: Nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. J. Virol. 1989, 63, 4653–4664. [Google Scholar] [CrossRef]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Spuul, P.; Salonen, A.; Merits, A.; Jokitalo, E.; Kaariainen, L.; Ahola, T. Role of the amphipathic peptide of Semliki forest virus replicase protein nsP1 in membrane association and virus replication. J. Virol. 2007, 81, 872–883. [Google Scholar] [CrossRef]
- Zusinaite, E.; Tints, K.; Kiiver, K.; Spuul, P.; Karo-Astover, L.; Merits, A.; Sarand, I. Mutations at the palmitoylation site of non-structural protein nsP1 of Semliki Forest virus attenuate virus replication and cause accumulation of compensatory mutations. J. Gen. Virol. 2007, 88, 1977–1985. [Google Scholar] [CrossRef]
- Pietila, M.K.; Hellstrom, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef]
- Coffey, L.L.; Beeharry, Y.; Borderia, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2021, 589, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Kaariainen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef]
- Ahola, T.; Lampio, A.; Auvinen, P.; Kaariainen, L. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J. 1999, 18, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Sawicki, S.G.; Sawicki, D.L. Sindbis virus nsP1 functions in negative-strand RNA synthesis. J. Virol. 1991, 65, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Hyvonen, M.; Peranen, J.; Kaariainen, L. Expression of Semliki Forest virus nsP1-specific methyltransferase in insect cells and in Escherichia coli. J. Virol. 1994, 68, 7418–7425. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Aher, P.P.; Lole, K.S. NTPase and 5'-RNA triphosphatase activities of Chikungunya virus nsP2 protein. PLoS ONE 2011, 6, e22336. [Google Scholar] [CrossRef]
- Rikkonen, M.; Peranen, J.; Kaariainen, L. ATPase and GTPase activities associated with Semliki Forest virus nonstructural protein nsP2. J. Virol. 1994, 68, 5804–5810. [Google Scholar] [CrossRef]
- Vasiljeva, L.; Merits, A.; Auvinen, P.; Kaariainen, L. Identification of a novel function of the alphavirus capping apparatus. RNA 5'-triphosphatase activity of Nsp2. J. Biol. Chem. 2000, 275, 17281–17287. [Google Scholar] [CrossRef] [PubMed]
- Gomez de Cedron, M.; Ehsani, N.; Mikkola, M.L.; Garcia, J.A.; Kaariainen, L. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett. 1999, 448, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Merits, A.; Vasiljeva, L.; Ahola, T.; Kaariainen, L.; Auvinen, P. Proteolytic processing of Semliki Forest virus-specific non-structural polyprotein by nsP2 protease. J. Gen. Virol. 2001, 82, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, D.; Barkhimer, D.B.; Sawicki, S.G.; Rice, C.M.; Schlesinger, S. Temperature sensitive shut-off of alphavirus minus strand RNA synthesis maps to a nonstructural protein, nsP4. Virology 1990, 174, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Ferguson, M.; Cordonin, C.; Fragkoudis, R.; Ool, M.; Tamberg, N.; Sherwood, K.; Fazakerley, J.K.; Merits, A. Differences in processing determinants of onstructural polyprotein and in the sequence of nonstructural protein 3 affect neurovirulence of Semliki Forest virus. J. Virol. 2015, 89, 11030–11045. [Google Scholar] [CrossRef] [PubMed]
- Tuittila, M.T.; Santagati, M.G.; Roytta, M.; Maatta, J.A.; Hinkkanen, A.E. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J. Virol. 2000, 74, 4579–4589. [Google Scholar] [CrossRef]
- Park, E.; Griffin, D.E. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef]
- McPherson, R.L.; Abraham, R.; Sreekumar, E.; Ong, S.E.; Cheng, S.J.; Baxter, V.K.; Kistemaker, H.A.; Filippov, D.V.; Griffin, D.E.; Leung, A.K. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc. Natl. Acad. Sci. USA 2017, 114, 1666–1671. [Google Scholar] [CrossRef]
- Meshram, C.D.; Agback, P.; Shiliaev, N.; Urakova, N.; Mobley, J.A.; Agback, T.; Frolova, E.I.; Frolov, I. Multiple host factors interact with hypervariable domain of chikungunya virus nsP3 and determine viral replication in cell-specific mode. J. Virol. 2018, 92, e00838-18. [Google Scholar] [CrossRef]
- Leung, A.K.; Vyas, S.; Rood, J.E.; Bhutkar, A.; Sharp, P.A.; Chang, P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell. 2011, 42, 489–499. [Google Scholar] [CrossRef]
- McInerney, G.M.; Kedersha, N.L.; Kaufman, R.J.; Anderson, P.; Liljestrom, P. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell. 2005, 16, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Jayabalan, A.K.; Adivarahan, S.; Koppula, A.; Abraham, R.; Batish, M.; Zenklusen, D.; Griffin, D.E.; Leung, A.K. Stress granule formation, disassembly, and composition are regulated by alphavirus ADP-ribosylhydrolase activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2021719118. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.; Hellstrom, K.; Jokitalo, E.; Ahola, T. RNA Replication and membrane modification require the same functions of alphavirus nonstructural proteins. J. Virol. 2015, 90, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Lemm, J.A.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [CrossRef] [PubMed]
- Shirako, Y.; Strauss, J.H. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994, 68, 1874–1885. [Google Scholar] [CrossRef]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Götte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G.; et al. Differential phosphatidylinositol-3-kinase-Akt-mTOR activation by Semliki Forest and chikungunya viruses is dependent on nsP3 and connected to replication complex internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef]
- Neuvonen, M.; Kazlauskas, A.; Martikainen, M.; Hinkkanen, A.; Ahola, T.; Saksela, K. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog. 2011, 7, e1002383. [Google Scholar] [CrossRef]
- Eckei, L.; Krieg, S.; Butepage, M.; Lehmann, A.; Gross, A.; Lippok, B.; Grimm, A.R.; Kümmerer, B.M.; Rossetti, G.; Lüscher, B.; et al. The conserved macrodomains of the non-structural proteins of Chikungunya virus and other pathogenic positive strand RNA viruses function as mono-ADP-ribosylhydrolases. Sci. Rep. 2017, 7, 41746. [Google Scholar] [CrossRef]
- Alhammad, Y.M.O.; Kashipathy, M.M.; Roy, A.; Gagné, J.P.; McDonald, P.; Gao, P.; Nonfoux, L.; Battaile, K.P.; Johnson, D.K.; Holmstrom, E.D.; et al. The SARS-CoV-2 conserved macrodomain is a mono-ADP-ribosylhydrolase. J. Virol. 2021, 95, e01969. [Google Scholar] [CrossRef]
- Luscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2022, 289, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Mikolcevic, P.; Mikoc, A.; Ahel, I. ADP-ribosylation signalling and human disease. Open Biol. 2019, 9, 190041. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Hauer, D.; McPherson, R.L.; Utt, A.; Kirby, I.T.; Cohen, M.S.; Merits, A.; Leung, A.K.; Griffin, D.E. ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, E10457–E10466. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral macro domains reverse protein ADP-ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef]
- Abraham, R.; McPherson, R.L.; Dasovich, M.; Badiee, M.; Leung, A.K.L.; Griffin, D.E. Both ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain affect neurovirulence in mice. mBio 2020, 11. [Google Scholar] [CrossRef]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during Severe Acute Respiratory Syndrome Coronavirus infection. mBio 2016, 7. [Google Scholar] [CrossRef]
- Grunewald, M.E.; Chen, Y.; Kuny, C.; Maejima, T.; Lease, R.; Ferraris, D.; Aikawa, M.; Sullivan, C.S.; Perlman, S.; Fehr, A.R. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019, 15, e1007756. [Google Scholar] [CrossRef]
- Malet, H.; Coutard, B.; Jamal, S.; Dutartre, H.; Papageorgiou, N.; Neuvonen, M.; Ahola, T.; Forrester, N.; Gould, E.A.; Lafitte, D.; et al. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol. 2009, 83, 6534–6545. [Google Scholar] [CrossRef]
- Jankevicius, G.; Hassler, M.; Golia, B.; Rybin, V.; Zacharias, M.; Timinszky, G.; Ladurner, A.G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struc. Mol. Biol. 2013, 20, 508–514. [Google Scholar] [CrossRef]
- Alliot, F.; Pessac, B. Astrocytic cell clones derived from established cultures of 8-day postnatal mouse cerebella. Brain Res. 1984, 306, 283–291. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Levitt, N.H.; Ramsburg, H.H.; Hasty, S.E.; Repik, P.M.; Cole Jr, F.E.; Lupton, H.W. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 1986, 4, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Gorchakov, R.; Wang, E.; Leal, G.; Forrester, N.L.; Plante, K.; Rossi, S.L.; Partidos, C.D.; Adams, A.P.; Seymour, R.L.; Weger, J.; et al. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J. Virol. 2012, 86, 6084–6096. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.S.; Singh, R.D.; Kim, K.K. Generation of a pure culture of neuron-like cells with a glutamatergic phenotype from mouse astrocytes. Biomedicines 2022, 10, 928. [Google Scholar] [CrossRef] [PubMed]
- Too, I.H.; Yeo, H.; Sessions, O.M.; Yan, B.; Libau, E.A.; Howe, J.L.; Lim, Z.Q.; Suku-Maran, S.; Ong, W.Y.; Chua, K.B.; et al. Enterovirus 71 infection of motor neuron-like NSC-34 cells undergoes a non-lytic exit pathway. Sci. Rep. 2016, 6, 36983. [Google Scholar] [CrossRef]
- Carrasco, L.; Sanz, M.A.; Gonzalez-Almela, E. The regulation of translation in alphavirus-infected cells. Viruses 2018, 10, 70. [Google Scholar] [CrossRef]
- Sokoloski, K.J.; Hayes, C.A.; Dunn, M.P.; Balke, J.L.; Hardy, R.W.; Mukhopadhyay, S. Sindbis virus infectivity improves during the course of infection in both mammalian and mosquito cells. Virus Res. 2012, 167, 26–33. [Google Scholar] [CrossRef]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef]
- Gotte, B.; Liu, L.; McInerney, G.M. The enigmatic alphavirus non-structural protein 3 (nsP3) revealing Its secrets at last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef]
- Carter-O'Connell, I.; Jin, H.; Morgan, R.K.; Zaja, R.; David, L.L.; Ahel, I.; Cohen, M.S. Identifying family-member-specific targets of mono-ARTDs by using a chemical genetics approach. Cell Rep. 2016, 14, 621–631. [Google Scholar] [CrossRef]
- Carter-O'Connell, I.; Vermehren-Schmaedick, A.; Jin, H.; Morgan, R.K.; David, L.L.; Cohen, M.S. Combining chemical genetics with proximity-dependent labeling reveals cellular targets of poly(ADP-ribose) polymerase 14 (PARP14). ACS Chem. Biol. 2018, 13, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Matkovic, R.; Bernard, E.; Fontanel, S.; Eldin, P.; Chazal, N.; Hassan Hersi, D.; Merits, A.; Péloponèse Jr, J.M.; Briant, L. The host DHX9 DExH-box helicase is recruited to chikungunya virus replication complexes for optimal genomic RNA rranslation. J. Virol. 2019, 93, e01764-18. [Google Scholar] [CrossRef]
- Gotte, B.; Panas, M.D.; Hellstrom, K.; Liu, L.; Samreen, B.; Larsson, O.; Ahola, T.; McInerney, G.M. Separate domains of G3BP promote efficient clustering of alphavirus replication complexes and recruitment of the translation initiation machinery. PLoS Pathog. 2019, 15, e1007842. [Google Scholar] [CrossRef] [PubMed]
- Scholte, F.E.; Tas, A.; Albulescu, I.C.; Žusinaite, E.; Merits, A.; Snijder, E.J.; van Hemert, M.J. Stress granule components G3BP1 and G3BP2 play a proviral role early in chikungunya virus replication. J. Virol. 2015, 89, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Reynaud, J.M.; Rasalouskaya, A.; Akhrymuk, I.; Mobley, J.A.; Frolov, I.; Frolova, E.I. New World and Old World alphaviruses have evolved to exploit different components of stress granules, FXR and G3BP proteins, for assembly of viral replication complexes. PLoS Pathog. 2016, 12, e1005810. [Google Scholar] [CrossRef]
- Kim, S.S.; Sze, L.; Liu, C.; Lam, K.P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-beta response. J. Biol. Chem. 2019, 294, 6430–6438. [Google Scholar] [CrossRef]
- Reineke, L.C.; Lloyd, R.E. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules. Curr. Biol. 2009, 19, R397–R398. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Lloyd, R.E. Cytoplasmic RNA granules and viral infection. Annu. Rev. Virol. 2014, 1, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip. Rev. RNA 2013, 4, 317–331. [Google Scholar] [CrossRef]
- Lindquist, M.E.; Lifland, A.W.; Utley, T.J.; Santangelo, P.J.; Crowe Jr, J.E. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J. Virol 2010, 84, 12274–12284. [Google Scholar] [CrossRef]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef]
- Zheng, Y.; Kielian, M. Imaging of the alphavirus capsid protein during virus replication. J. Virol. 2013, 87, 9579–9589. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Kedersha, N.; McInerney, G.M. Methods for the characterization of stress granules in virus infected cells. Methods 2015, 90, 57–64. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Burnham, A.J.; Gong, L.; Hardy, R.W. Heterogeneous nuclear ribonuclear protein K interacts.with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 2007, 367, 212–221. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, A.T.; Gebhart, N.N.; Meller, M.E.; Hardy, R.W.; Sokoloski, K.J. Identification and Characterization of Sindbis Virus RNA-Host Protein Interactions. J. Virol. 2018, 92, e02171-17. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res. 2009, 37, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. Poly(ADP-Ribosyl)ation of hnRNP A1 Protein Controls Translational Repression in Drosophila. Mol. Cell Biol. 2016, 36, 2476–2486. [Google Scholar] [CrossRef]
- Sahoo, P.K.; Lee, S.J.; Jaiswal, P.B.; Alber, S.; Kar, A.N.; Miller-Randolph, S.; Taylor, E.E.; Smith, T.; Singh, B.; Ho, T.S.Y.; et al. Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Comm. 2018, 9, 3358. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.D.; Willers, I.M.; Sala, S.; Cuezva, J.M. Human G3BP1 interacts with beta-F1-ATPase mRNA and inhibits its translation. J. Cell Sci. 2010, 123, 2685–2696. [Google Scholar] [CrossRef]
- Bidet, K.; Dadlani, D.; Garcia-Blanco, M.A. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog. 2014, 10, e1004242. [Google Scholar] [CrossRef]
- Gagne, J.P.; Hunter, J.M.; Labrecque, B.; Chabot, B.; Poirier, G.G. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem. J. 2003, 371, 331–340. [Google Scholar] [CrossRef]
- Westcott, C.E.; Qazi, S.; Maiocco, A.M.; Mukhopadhyay, S.; Sokoloski, K.J. Binding of hnRNP I-vRNA regulates Sindbis virus structural protein expression to promote particle infectivity. Viruses 2022, 14, 1423. [Google Scholar] [CrossRef]
- Yeh, J.X.; Fan, Y.; Bartlett, M.L.; Zhang, X.; Sadowski, N.; Hauer, D.A.; Timp, W.; Griffin, D.E. Treatment of Sindbis virus-infected neurons with antibody to E2 alters synthesis of complete and nsP1-expressing defective viral RNAs. mBio 2022, 13, e0222122. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.; Abraham, R.; Pieterse, L.; Yeh, J.X.; Griffin, D.E. Cell-Type-Dependent Role for nsP3 Macrodomain ADP-Ribose Binding and Hydrolase Activity during Chikungunya Virus Infection. Viruses 2022, 14, 2744. https://doi.org/10.3390/v14122744
Kim T, Abraham R, Pieterse L, Yeh JX, Griffin DE. Cell-Type-Dependent Role for nsP3 Macrodomain ADP-Ribose Binding and Hydrolase Activity during Chikungunya Virus Infection. Viruses. 2022; 14(12):2744. https://doi.org/10.3390/v14122744
Chicago/Turabian StyleKim, Taewoo, Rachy Abraham, Lisa Pieterse, Jane X. Yeh, and Diane E. Griffin. 2022. "Cell-Type-Dependent Role for nsP3 Macrodomain ADP-Ribose Binding and Hydrolase Activity during Chikungunya Virus Infection" Viruses 14, no. 12: 2744. https://doi.org/10.3390/v14122744
APA StyleKim, T., Abraham, R., Pieterse, L., Yeh, J. X., & Griffin, D. E. (2022). Cell-Type-Dependent Role for nsP3 Macrodomain ADP-Ribose Binding and Hydrolase Activity during Chikungunya Virus Infection. Viruses, 14(12), 2744. https://doi.org/10.3390/v14122744