
Different Pathways Conferring Integrase Strand-Transfer Inhibitors Resistance


Abstract
1. Introduction
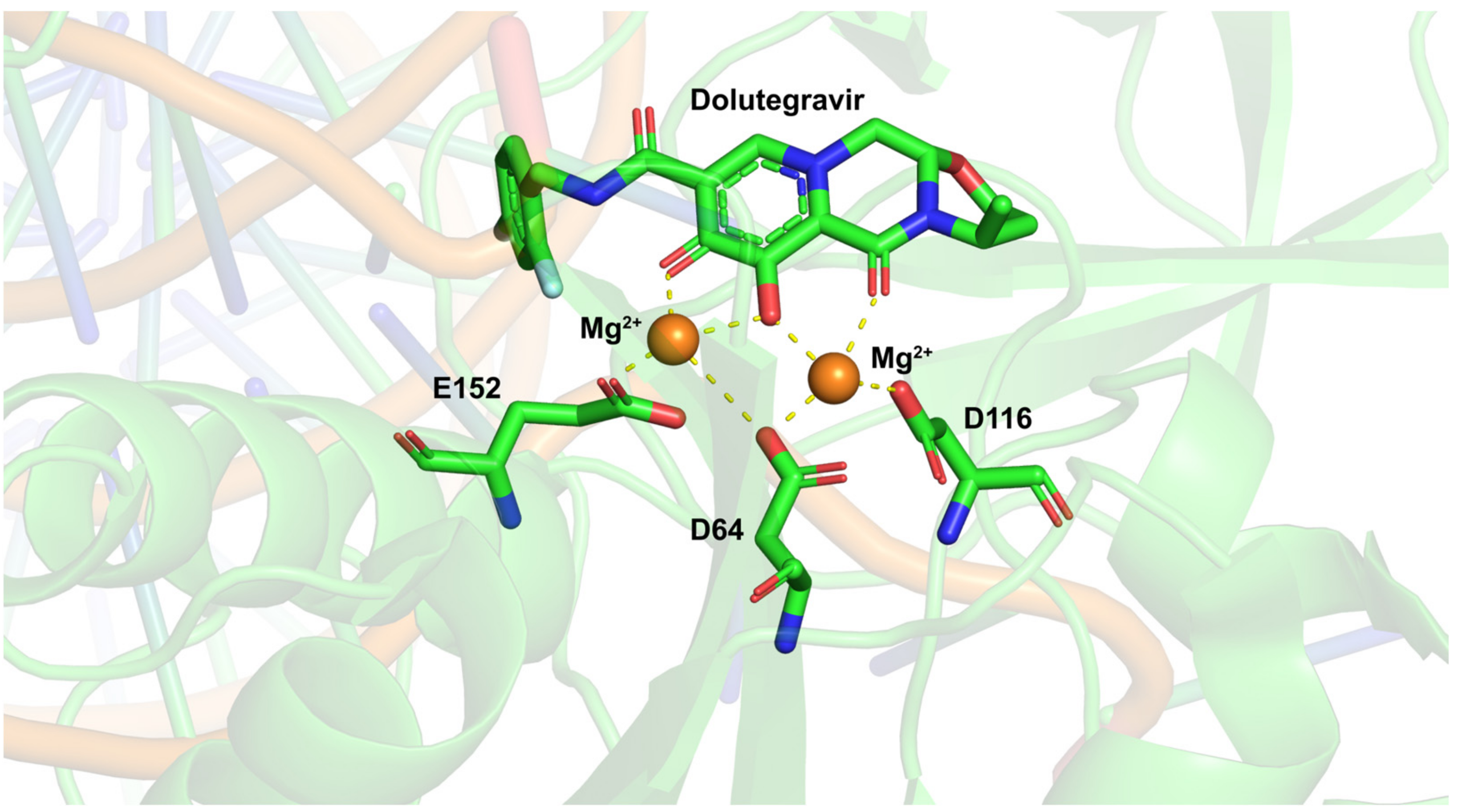
2. Integrase Function Involved in Viral Integration
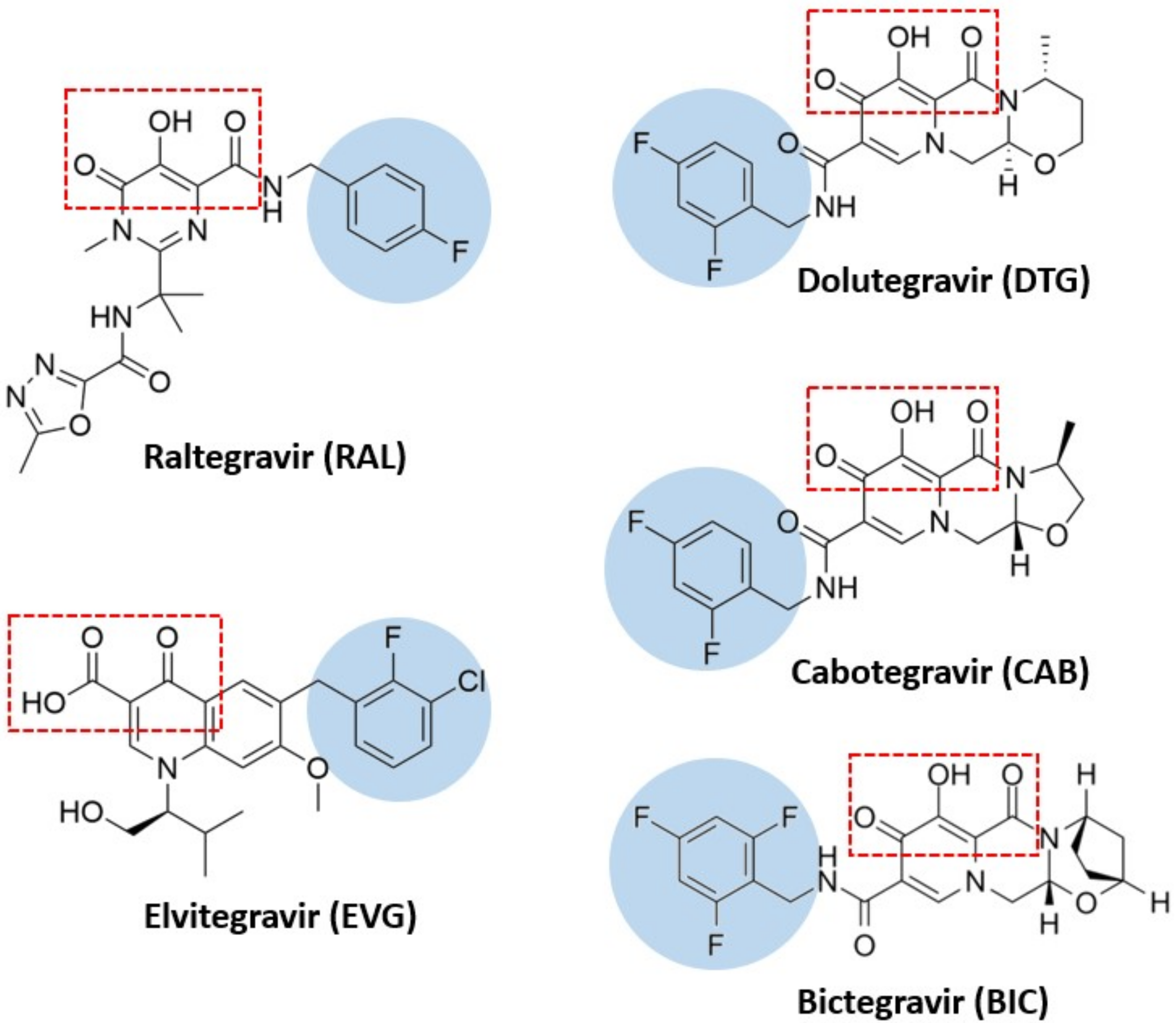
3. Inhibitors
3.1. Raltegravir
3.2. Elvitegravir
3.3. Dolutegravir

3.4. Bictegravir and Cabotegravir
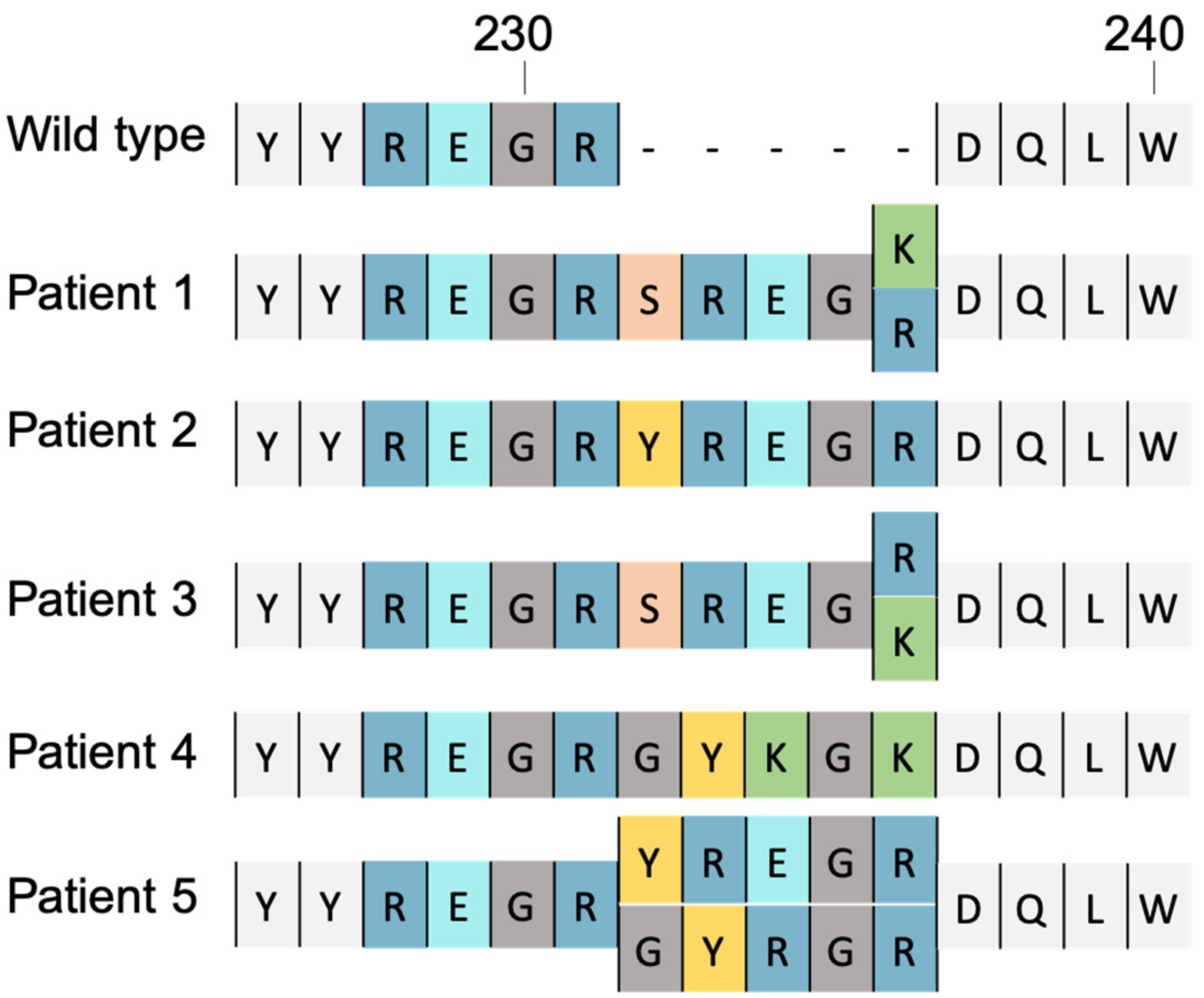
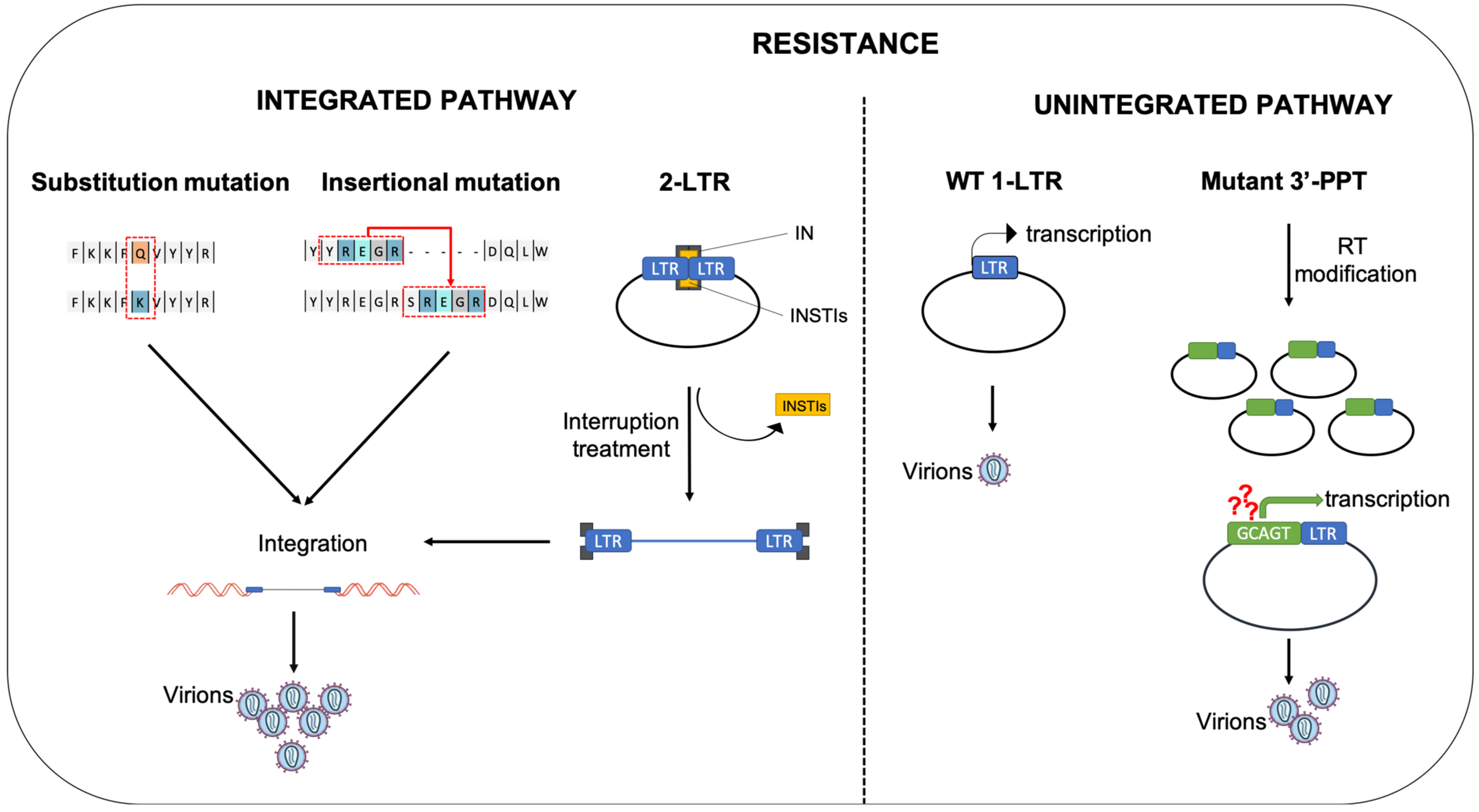
4. Mutations Leading to Resistance
4.1. Substitution Mutation
4.2. Insertional Mutation
5. Role of Unintegrated Viral DNA in Resistance to INSTIs
5.1. The 2-LTR Circles, a Reservoir of HIV-1 Genomes
5.2. Expression of uDNA
5.3. Mutations in the 3′-PPT: A Way to Escape from INSTIs Using uDNA
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, E.M.; Hope, T.J. HIV-1 Capsid: The Multifaceted Key Player in HIV-1 Infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Crumpacker, C. HIV UTR, LTR, and Epigenetic Immunity. Viruses 2022, 14, 1084. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.O.; Bowerman, B.; Varmus, H.E.; Bishop, J.M. Correct Integration of Retroviral DNA in Vitro. Cell 1987, 49, 347–356. [Google Scholar] [CrossRef]
- Hu, W.-S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human Immunodeficiency Virus Type 1 Preintegration Complexes: Studies of Organization and Composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA Integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef]
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV-1 DNA Integration: Mechanism of Viral DNA Cleavage and DNA Strand Transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef]
- Gerton, J.L.; Brown, P.O. The Core Domain of HIV-1 Integrase Recognizes Key Features of Its DNA Substrates. J. Biol. Chem. 1997, 272, 25809–25815. [Google Scholar] [CrossRef]
- Laboulais, C.; Deprez, E.; Leh, H.; Mouscadet, J.F.; Brochon, J.C.; Le Bret, M. HIV-1 Integrase Catalytic Core: Molecular Dynamics and Simulated Fluorescence Decays. Biophys. J. 2001, 81, 473–489. [Google Scholar] [CrossRef]
- Delelis, O.; Parissi, V.; Leh, H.; Mbemba, G.; Petit, C.; Sonigo, P.; Deprez, E.; Mouscadet, J.-F. Efficient and Specific Internal Cleavage of a Retroviral Palindromic DNA Sequence by Tetrameric HIV-1 Integrase. PLoS ONE 2007, 2, e608. [Google Scholar] [CrossRef]
- Delelis, O.; Petit, C.; Leh, H.; Mbemba, G.; Mouscadet, J.-F.; Sonigo, P. A Novel Function for Spumaretrovirus Integrase: An Early Requirement for Integrase-Mediated Cleavage of 2 LTR Circles. Retrovirology 2005, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Thierry, S.; Munir, S.; Thierry, E.; Subra, F.; Leh, H.; Zamborlini, A.; Saenz, D.; Levy, D.N.; Lesbats, P.; Saïb, A.; et al. Integrase Inhibitor Reversal Dynamics Indicate Unintegrated HIV-1 Dna Initiate de Novo Integration. Retrovirology 2015, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 Integrase Forms Stable Tetramers and Associates with LEDGF/P75 Protein in Human Cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.; Calmels, C.; Desjobert, C.; Castroviejo, M.; Caumont-Sarcos, A.; Tarrago-Litvak, L.; Litvak, S.; Parissi, V. HIV-1 Integrase Crosslinked Oligomers Are Active in Vitro. Nucleic Acids Res. 2005, 33, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Di Nunzio, F.; Labeja, A.; Wang, J.; Engelman, A.; Cherepanov, P. Structural Basis for Functional Tetramerization of Lentiviral Integrase. PLoS Pathog. 2009, 5, e1000515. [Google Scholar] [CrossRef]
- Saag, M.S.; Benson, C.A.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Mugavero, M.J.; Sax, P.E.; Smith, D.M.; Thompson, M.A.; Buchbinder, S.P.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2018 Recommendations of the International Antiviral Society-USA Panel. JAMA 2018, 320, 379–396. [Google Scholar] [CrossRef]
- Svarovskaia, E.S.; Barr, R.; Zhang, X.; Pais, G.C.G.; Marchand, C.; Pommier, Y.; Burke, T.R.; Pathak, V.K. Azido-Containing Diketo Acid Derivatives Inhibit Human Immunodeficiency Virus Type 1 Integrase in Vivo and Influence the Frequency of Deletions at Two-Long-Terminal-Repeat-Circle Junctions. J. Virol. 2004, 78, 3210–3222. [Google Scholar] [CrossRef]
- Kilzer, J.M.; Stracker, T.; Beitzel, B.; Meek, K.; Weitzman, M.; Bushman, F.D. Roles of Host Cell Factors in Circularization of Retroviral Dna. Virology 2003, 314, 460–467. [Google Scholar] [CrossRef]
- Hindmarsh, P.; Leis, J. Retroviral DNA Integration. Microbiol. Mol. Biol. Rev. MMBR 1999, 63, 836–843. [Google Scholar] [CrossRef]
- Engelman, A.N.; Kvaratskhelia, M. Multimodal Functionalities of HIV-1 Integrase. Viruses 2022, 14, 926. [Google Scholar] [CrossRef]
- Zheng, R.; Jenkins, T.M.; Craigie, R. Zinc Folds the N-Terminal Domain of HIV-1 Integrase, Promotes Multimerization, and Enhances Catalytic Activity. Proc. Natl. Acad. Sci. USA 1996, 93, 13659–13664. [Google Scholar] [CrossRef] [PubMed]
- Carayon, K.; Leh, H.; Henry, E.; Simon, F.; Mouscadet, J.-F.; Deprez, E. A Cooperative and Specific DNA-Binding Mode of HIV-1 Integrase Depends on the Nature of the Metallic Cofactor and Involves the Zinc-Containing N-Terminal Domain. Nucleic Acids Res. 2010, 38, 3692–3708. [Google Scholar] [CrossRef]
- Goldgur, Y.; Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Craigie, R.; Davies, D.R. Three New Structures of the Core Domain of HIV-1 Integrase: An Active Site That Binds Magnesium. Proc. Natl. Acad. Sci. USA 1998, 95, 9150–9154. [Google Scholar] [CrossRef]
- Maignan, S.; Guilloteau, J.P.; Zhou-Liu, Q.; Clément-Mella, C.; Mikol, V. Crystal Structures of the Catalytic Domain of HIV-1 Integrase Free and Complexed with Its Metal Cofactor: High Level of Similarity of the Active Site with Other Viral Integrases. J. Mol. Biol. 1998, 282, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Lutzke, R.A.; Plasterk, R.H. Structure-Based Mutational Analysis of the C-Terminal DNA-Binding Domain of Human Immunodeficiency Virus Type 1 Integrase: Critical Residues for Protein Oligomerization and DNA Binding. J. Virol. 1998, 72, 4841–4848. [Google Scholar] [CrossRef]
- Maertens, G.N.; Engelman, A.N.; Cherepanov, P. Structure and Function of Retroviral Integrase. Nat. Rev. Microbiol. 2022, 20, 20–34. [Google Scholar] [CrossRef]
- Pflieger, A.; Waffo Teguo, P.; Papastamoulis, Y.; Chaignepain, S.; Subra, F.; Munir, S.; Delelis, O.; Lesbats, P.; Calmels, C.; Andreola, M.-L.; et al. Natural Stilbenoids Isolated from Grapevine Exhibiting Inhibitory Effects against HIV-1 Integrase and Eukaryote MOS1 Transposase in Vitro Activities. PLoS ONE 2013, 8, e81184. [Google Scholar] [CrossRef]
- Cosnefroy, O.; Tocco, A.; Lesbats, P.; Thierry, S.; Calmels, C.; Wiktorowicz, T.; Reigadas, S.; Kwon, Y.; De Cian, A.; Desfarges, S.; et al. Stimulation of the Human RAD51 Nucleofilament Restricts HIV-1 Integration in Vitro and in Infected Cells. J. Virol. 2012, 86, 513–526. [Google Scholar] [CrossRef]
- Deprez, E.; Tauc, P.; Leh, H.; Mouscadet, J.F.; Auclair, C.; Hawkins, M.E.; Brochon, J.C. DNA Binding Induces Dissociation of the Multimeric Form of HIV-1 Integrase: A Time-Resolved Fluorescence Anisotropy Study. Proc. Natl. Acad. Sci. USA 2001, 98, 10090–10095. [Google Scholar] [CrossRef]
- Delelis, O.; Carayon, K.; Guiot, E.; Leh, H.; Tauc, P.; Brochon, J.-C.; Mouscadet, J.-F.; Deprez, E. Insight into the Integrase-DNA Recognition Mechanism. A Specific DNA-Binding Mode Revealed by an Enzymatically Labeled Integrase. J. Biol. Chem. 2008, 283, 27838–27849. [Google Scholar] [CrossRef]
- Li, M.; Mizuuchi, M.; Burke, T.R.; Craigie, R. Retroviral DNA Integration: Reaction Pathway and Critical Intermediates. EMBO J. 2006, 25, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Craigie, R. Processing of Viral DNA Ends Channels the HIV-1 Integration Reaction to Concerted Integration. J. Biol. Chem. 2005, 280, 29334–29339. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, S.; Mousnier, A.; Busschots, K.; Maroun, M.; Van Maele, B.; Tempé, D.; Vandekerckhove, L.; Moisant, F.; Ben-Slama, L.; Witvrouw, M.; et al. Integrase Mutants Defective for Interaction with LEDGF/P75 Are Impaired in Chromosome Tethering and HIV-1 Replication. J. Biol. Chem. 2005, 280, 25517–25523. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Balakrishnan, M.; Jonsson, C.B. Major and Minor Groove Contacts in Retroviral Integrase-LTR Interactions. Biochemistry 1999, 38, 3624–3632. [Google Scholar] [CrossRef]
- Taganov, K.D.; Cuesta, I.; Daniel, R.; Cirillo, L.A.; Katz, R.A.; Zaret, K.S.; Skalka, A.M. Integrase-Specific Enhancement and Suppression of Retroviral DNA Integration by Compacted Chromatin Structure in Vitro. J. Virol. 2004, 78, 5848–5855. [Google Scholar] [CrossRef]
- Deprez, E.; Barbe, S.; Kolaski, M.; Leh, H.; Zouhiri, F.; Auclair, C.; Brochon, J.-C.; Le Bret, M.; Mouscadet, J.-F. Mechanism of HIV-1 Integrase Inhibition by Styrylquinoline Derivatives in Vitro. Mol. Pharmacol. 2004, 65, 85–98. [Google Scholar] [CrossRef]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of Strand Transfer That Prevent Integration and Inhibit HIV-1 Replication in Cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef]
- Marchand, C.; Zhang, X.; Pais, G.C.G.; Cowansage, K.; Neamati, N.; Burke, T.R.; Pommier, Y. Structural Determinants for HIV-1 Integrase Inhibition by Beta-Diketo Acids. J. Biol. Chem. 2002, 277, 12596–12603. [Google Scholar] [CrossRef]
- Espeseth, A.S.; Felock, P.; Wolfe, A.; Witmer, M.; Grobler, J.; Anthony, N.; Egbertson, M.; Melamed, J.Y.; Young, S.; Hamill, T.; et al. HIV-1 Integrase Inhibitors That Compete with the Target DNA Substrate Define a Unique Strand Transfer Conformation for Integrase. Proc. Natl. Acad. Sci. USA 2000, 97, 11244–11249. [Google Scholar] [CrossRef]
- Grobler, J.A.; Stillmock, K.; Hu, B.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J.; et al. Diketo Acid Inhibitor Mechanism and HIV-1 Integrase: Implications for Metal Binding in the Active Site of Phosphotransferase Enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 6661–6666. [Google Scholar] [CrossRef]
- Malet, I.; Delelis, O.; Valantin, M.-A.; Montes, B.; Soulie, C.; Wirden, M.; Tchertanov, L.; Peytavin, G.; Reynes, J.; Mouscadet, J.-F.; et al. Mutations Associated with Failure of Raltegravir Treatment Affect Integrase Sensitivity to the Inhibitor in Vitro. Antimicrob. Agents Chemother. 2008, 52, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Steigbigel, R.T.; Cooper, D.A.; Kumar, P.N.; Eron, J.E.; Schechter, M.; Markowitz, M.; Loutfy, M.R.; Lennox, J.L.; Gatell, J.M.; Rockstroh, J.K.; et al. Raltegravir with Optimized Background Therapy for Resistant HIV-1 Infection. N. Engl. J. Med. 2008, 359, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Nguyen, B.-Y.; Gotuzzo, E.; Mendo, F.; Ratanasuwan, W.; Kovacs, C.; Prada, G.; Morales-Ramirez, J.O.; Crumpacker, C.S.; Isaacs, R.D.; et al. Rapid and Durable Antiretroviral Effect of the HIV-1 Integrase Inhibitor Raltegravir as Part of Combination Therapy in Treatment-Naive Patients with HIV-1 Infection: Results of a 48-Week Controlled Study. J. Acquir. Immune Defic. Syndr. 2007, 46, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rockstroh, J.K.; DeJesus, E.; Lennox, J.L.; Yazdanpanah, Y.; Saag, M.S.; Wan, H.; Rodgers, A.J.; Walker, M.L.; Miller, M.; DiNubile, M.J.; et al. Durable Efficacy and Safety of Raltegravir versus Efavirenz When Combined with Tenofovir/Emtricitabine in Treatment-Naive HIV-1-Infected Patients: Final 5-Year Results from STARTMRK. J. Acquir. Immune Defic. Syndr. 1999 2013, 63, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Hightower, K.E.; Wang, R.; Deanda, F.; Johns, B.A.; Weaver, K.; Shen, Y.; Tomberlin, G.H.; Carter, H.L.; Broderick, T.; Sigethy, S.; et al. Dolutegravir (S/GSK1349572) Exhibits Significantly Slower Dissociation than Raltegravir and Elvitegravir from Wild-Type and Integrase Inhibitor-Resistant HIV-1 Integrase-DNA Complexes. Antimicrob. Agents Chemother. 2011, 55, 4552–4559. [Google Scholar] [CrossRef]
- White, K.L.; Osman, N.; Cuadra-Foy, E.; Brenner, B.G.; Shivakumar, D.; Campigotto, F.; Tsiang, M.; Morganelli, P.A.; Novikov, N.; Lazerwith, S.E.; et al. Long Dissociation of Bictegravir from HIV-1 Integrase-DNA Complexes. Antimicrob. Agents Chemother. 2021, 65, e02406-20. [Google Scholar] [CrossRef]
- Imaz, A.; Podzamczer, D. Tenofovir Alafenamide, Emtricitabine, Elvitegravir, and Cobicistat Combination Therapy for the Treatment of HIV. Expert Rev. Anti Infect. Ther. 2017, 15, 195–209. [Google Scholar] [CrossRef]
- Marinello, J.; Marchand, C.; Mott, B.T.; Bain, A.; Thomas, C.J.; Pommier, Y. Comparison of Raltegravir and Elvitegravir on HIV-1 Integrase Catalytic Reactions and on a Series of Drug-Resistant Integrase Mutants. Biochemistry 2008, 47, 9345–9354. [Google Scholar] [CrossRef]
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV Drug Resistance against Strand Transfer Integrase Inhibitors. Retrovirology 2017, 14, 36. [Google Scholar] [CrossRef]
- Greener, B.N.; Patterson, K.B.; Prince, H.M.A.; Sykes, C.S.; Adams, J.L.; Dumond, J.B.; Shaheen, N.J.; Madanick, R.D.; Dellon, E.S.; Cohen, M.S.; et al. Dolutegravir Pharmacokinetics in the Genital Tract and Colorectum of HIV-Negative Men after Single and Multiple Dosing. J. Acquir. Immune Defic. Syndr. 2013, 64, 39–44. [Google Scholar] [CrossRef]
- Eron, J.J.; Clotet, B.; Durant, J.; Katlama, C.; Kumar, P.; Lazzarin, A.; Poizot-Martin, I.; Richmond, G.; Soriano, V.; Ait-Khaled, M.; et al. Safety and Efficacy of Dolutegravir in Treatment-Experienced Subjects with Raltegravir-Resistant HIV Type 1 Infection: 24-Week Results of the VIKING Study. J. Infect. Dis. 2013, 207, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Dow, D.E.; Bartlett, J.A. Dolutegravir, the Second-Generation of Integrase Strand Transfer Inhibitors (INSTIs) for the Treatment of HIV. Infect. Dis. Ther. 2014, 3, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, X.; Wang, H.; Jurado, K.A.; Engelman, A.N.; Craigie, R. A Peptide Derived from Lens Epithelium–Derived Growth Factor Stimulates HIV-1 DNA Integration and Facilitates Intasome Structural Studies. J. Mol. Biol. 2020, 432, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Discovery of Bictegravir (GS-9883), a Novel, Unboosted, Once-Daily HIV-1 Integrase Strand Transfer Inhibitor (INSTI) with Improved Pharmacokinetics and In Vitro Resistance Profile. Available online: https://www.natap.org/2016/HIV/062016_05.htm (accessed on 21 September 2022).
- Yoshinaga, T.; Kobayashi, M.; Seki, T.; Miki, S.; Wakasa-Morimoto, C.; Suyama-Kagitani, A.; Kawauchi-Miki, S.; Taishi, T.; Kawasuji, T.; Johns, B.A.; et al. Antiviral Characteristics of GSK1265744, an HIV Integrase Inhibitor Dosed Orally or by Long-Acting Injection. Antimicrob. Agents Chemother. 2015, 59, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Tsiang, M.; Jones, G.S.; Goldsmith, J.; Mulato, A.; Hansen, D.; Kan, E.; Tsai, L.; Bam, R.A.; Stepan, G.; Stray, K.M.; et al. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob. Agents Chemother. 2016, 60, 7086–7097. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors 3. A Diastereomeric Approach to Chiral Nonracemic Tricyclic Ring Systems and the Discovery of Dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef]
- Han, Y.; Mesplède, T.; Wainberg, M.A. Investigational HIV Integrase Inhibitors in Phase I and Phase II Clinical Trials. Expert Opin. Investig. Drugs 2017, 26, 1207–1213. [Google Scholar] [CrossRef]
- McPherson, T.D.; Sobieszczyk, M.E.; Markowitz, M. Cabotegravir in the Treatment and Prevention of Human Immunodeficiency Virus-1. Expert Opin. Investig. Drugs 2018, 27, 413–420. [Google Scholar] [CrossRef]
- Ford, S.L.; Gould, E.; Chen, S.; Margolis, D.; Spreen, W.; Crauwels, H.; Piscitelli, S. Lack of Pharmacokinetic Interaction between Rilpivirine and Integrase Inhibitors Dolutegravir and GSK1265744. Antimicrob. Agents Chemother. 2013, 57, 5472–5477. [Google Scholar] [CrossRef]
- Spreen, W.; Min, S.; Ford, S.L.; Chen, S.; Lou, Y.; Bomar, M.; St Clair, M.; Piscitelli, S.; Fujiwara, T. Pharmacokinetics, Safety, and Monotherapy Antiviral Activity of GSK1265744, an HIV Integrase Strand Transfer Inhibitor. HIV Clin. Trials 2013, 14, 192–203. [Google Scholar] [CrossRef]
- Marcelin, A.-G.; Ceccherini-Silberstein, F.; Perno, C.-F.; Calvez, V. Resistance to Novel Drug Classes. Curr. Opin. HIV AIDS 2009, 4, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.A.; Calvez, V.; Günthard, H.F.; Paredes, R.; Pillay, D.; Shafer, R.; Wensing, A.M.; Richman, D.D. 2011 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2011, 19, 156–164. [Google Scholar] [PubMed]
- Charpentier, C.; Roquebert, B.; Delelis, O.; Larrouy, L.; Matheron, S.; Tubiana, R.; Karmochkine, M.; Duval, X.; Chêne, G.; Storto, A.; et al. Hot Spots of Integrase Genotypic Changes Leading to HIV-2 Resistance to Raltegravir. Antimicrob. Agents Chemother. 2011, 55, 1293–1295. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.A.; Mesplède, T.; Quashie, P.K. The Development of Novel HIV Integrase Inhibitors and the Problem of Drug Resistance. Curr. Opin. Virol. 2012, 2, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Orrell, C.; Hagins, D.P.; Belonosova, E.; Porteiro, N.; Walmsley, S.; Falcó, V.; Man, C.Y.; Aylott, A.; Buchanan, A.M.; Wynne, B.; et al. Fixed-Dose Combination Dolutegravir, Abacavir, and Lamivudine versus Ritonavir-Boosted Atazanavir plus Tenofovir Disoproxil Fumarate and Emtricitabine in Previously Untreated Women with HIV-1 Infection (ARIA): Week 48 Results from a Randomised, Open-Label, Non-Inferiority, Phase 3b Study. Lancet HIV 2017, 4, e536–e546. [Google Scholar] [CrossRef]
- Oliveira, M.; Mesplède, T.; Moïsi, D.; Ibanescu, R.-I.; Brenner, B.; Wainberg, M.A. The Dolutegravir R263K Resistance Mutation in HIV-1 Integrase Is Incompatible with the Emergence of Resistance against Raltegravir. AIDS Lond. Engl. 2015, 29, 2255–2260. [Google Scholar] [CrossRef]
- Quashie, P.K.; Oliviera, M.; Veres, T.; Osman, N.; Han, Y.-S.; Hassounah, S.; Lie, Y.; Huang, W.; Mesplède, T.; Wainberg, M.A. Differential Effects of the G118R, H51Y, and E138K Resistance Substitutions in Different Subtypes of HIV Integrase. J. Virol. 2015, 89, 3163–3175. [Google Scholar] [CrossRef]
- Le Hingrat, Q.; Collin, G.; Lê, M.; Peytavin, G.; Visseaux, B.; Bertine, M.; Tubiana, R.; Karmochkine, M.; Valin, N.; Collin, F.; et al. A New Mechanism of Resistance of Human Immunodeficiency Virus Type 2 to Integrase Inhibitors: A 5-Amino-Acid Insertion in the Integrase C-Terminal Domain. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2019, 69, 657–667. [Google Scholar] [CrossRef]
- Pham, H.T.; Hassounah, S.; Keele, B.F.; Van Rompay, K.K.A.; Mesplède, T. Insertion as a Resistance Mechanism Against Integrase Inhibitors in Several Retroviruses. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2019, 69, 1460–1461. [Google Scholar] [CrossRef]
- Pereira-Vaz, J.; Crespo, P.; Mocho, L.; Martinho, P.; Fidalgo, T.; Correia, L.; Rodrigues, F.; Duque, V. Identification of a New 2-Amino Acid Insertion in the Integrase Coding Region of HIV-1 Subtype G Isolates. J. Med. Virol. 2021, 93, 6388–6392. [Google Scholar] [CrossRef]
- Molina, J.-M.; Clotet, B.; van Lunzen, J.; Lazzarin, A.; Cavassini, M.; Henry, K.; Kulagin, V.; Givens, N.; de Oliveira, C.F.; Brennan, C.; et al. Once-Daily Dolutegravir versus Darunavir plus Ritonavir for Treatment-Naive Adults with HIV-1 Infection (FLAMINGO): 96 Week Results from a Randomised, Open-Label, Phase 3b Study. Lancet HIV 2015, 2, e127–e136. [Google Scholar] [CrossRef]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of Latent Tissue Reservoirs and Total Body Viral Load in HIV-1 Infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Munir, S.; Thierry, S.; Subra, F.; Deprez, E.; Delelis, O. Quantitative Analysis of the Time-Course of Viral DNA Forms during the HIV-1 Life Cycle. Retrovirology 2013, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, M.E.; Teo, I.; Greenough, T.; Sharova, N.; Luzuriaga, K.; Sullivan, J.L.; Bucy, R.P.; Kostrikis, L.G.; Haase, A.; Veryard, C.; et al. Persistence of Episomal HIV-1 Infection Intermediates in Patients on Highly Active Anti-Retroviral Therapy. Nat. Med. 2000, 6, 76–81. [Google Scholar] [CrossRef]
- Lai, M.; Maori, E.; Quaranta, P.; Matteoli, G.; Maggi, F.; Sgarbanti, M.; Crucitta, S.; Pacini, S.; Turriziani, O.; Antonelli, G.; et al. CRISPR/Cas9 Ablation of Integrated HIV-1 Accumulates Proviral DNA Circles with Reformed Long Terminal Repeats. J. Virol. 2021, 95, e0135821. [Google Scholar] [CrossRef]
- Farnet, C.M.; Haseltine, W.A. Circularization of Human Immunodeficiency Virus Type 1 DNA in Vitro. J. Virol. 1991, 65, 6942–6952. [Google Scholar] [CrossRef]
- Jeanson, L.; Subra, F.; Vaganay, S.; Hervy, M.; Marangoni, E.; Bourhis, J.; Mouscadet, J.-F. Effect of Ku80 Depletion on the Preintegrative Steps of HIV-1 Replication in Human Cells. Virology 2002, 300, 100–108. [Google Scholar] [CrossRef]
- Li, L.; Olvera, J.M.; Yoder, K.E.; Mitchell, R.S.; Butler, S.L.; Lieber, M.; Martin, S.L.; Bushman, F.D. Role of the Non-Homologous DNA End Joining Pathway in the Early Steps of Retroviral Infection. EMBO J. 2001, 20, 3272–3281. [Google Scholar] [CrossRef]
- Miller, M.D.; Wang, B.; Bushman, F.D. Human Immunodeficiency Virus Type 1 Preintegration Complexes Containing Discontinuous plus Strands Are Competent to Integrate in Vitro. J. Virol. 1995, 69, 3938–3944. [Google Scholar] [CrossRef]
- Sharkey, M.; Triques, K.; Kuritzkes, D.R.; Stevenson, M. In Vivo Evidence for Instability of Episomal Human Immunodeficiency Virus Type 1 CDNA. J. Virol. 2005, 79, 5203–5210. [Google Scholar] [CrossRef]
- Zhu, W.; Jiao, Y.; Lei, R.; Hua, W.; Wang, R.; Ji, Y.; Liu, Z.; Wei, F.; Zhang, T.; Shi, X.; et al. Rapid Turnover of 2-LTR HIV-1 DNA during Early Stage of Highly Active Antiretroviral Therapy. PLoS ONE 2011, 6, e21081. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Butler, S.L.; Johnson, E.P.; Bushman, F.D. Human Immunodeficiency Virus CDNA Metabolism: Notable Stability of Two-Long Terminal Repeat Circles. J. Virol. 2002, 76, 3739–3747. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Gange, S.J.; Siliciano, R.F. Intrinsic Stability of Episomal Circles Formed during Human Immunodeficiency Virus Type 1 Replication. J. Virol. 2002, 76, 4138–4144. [Google Scholar] [CrossRef] [PubMed]
- Gillim-Ross, L.; Cara, A.; Klotman, M.E. HIV-1 Extrachromosomal 2-LTR Circular DNA Is Long-Lived in Human Macrophages. Viral Immunol. 2005, 18, 190–196. [Google Scholar] [CrossRef]
- Pace, M.J.; Graf, E.H.; O’Doherty, U. HIV 2-Long Terminal Repeat Circular DNA Is Stable in Primary CD4+T Cells. Virology 2013, 441, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Muñoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective Gene Therapy with Nonintegrating Lentiviral Vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef]
- Philippe, S.; Sarkis, C.; Barkats, M.; Mammeri, H.; Ladroue, C.; Petit, C.; Mallet, J.; Serguera, C. Lentiviral Vectors with a Defective Integrase Allow Efficient and Sustained Transgene Expression in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 17684–17689. [Google Scholar] [CrossRef]
- Sloan, R.D.; Wainberg, M.A. The Role of Unintegrated DNA in HIV Infection. Retrovirology 2011, 8, 52. [Google Scholar] [CrossRef]
- Zhang, D.; He, H.; Guo, S. Hairpin DNA Probe-Based Fluorescence Assay for Detecting Palindrome Cleavage Activity of HIV-1 Integrase. Anal. Biochem. 2014, 460, 36–38. [Google Scholar] [CrossRef]
- Richetta, C.; Thierry, S.; Thierry, E.; Lesbats, P.; Lapaillerie, D.; Munir, S.; Subra, F.; Leh, H.; Deprez, E.; Parissi, V.; et al. Two-Long Terminal Repeat (LTR) DNA Circles Are a Substrate for HIV-1 Integrase. J. Biol. Chem. 2019, 294, 8286–8295. [Google Scholar] [CrossRef]
- Goff, S.P. Silencing of Unintegrated Retroviral DNAs. Viruses 2021, 13, 2248. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Haggerty, S.; Lamonica, C.A.; Meier, C.M.; Welch, S.K.; Wasiak, A.J. Integration Is Not Necessary for Expression of Human Immunodeficiency Virus Type 1 Protein Products. J. Virol. 1990, 64, 2421–2425. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.R.; Yu, D.; Biancotto, A.; Margolis, L.B.; Wu, Y. Measurement of Human Immunodeficiency Virus Type 1 Preintegration Transcription by Using Rev-Dependent Rev-CEM Cells Reveals a Sizable Transcribing DNA Population Comparable to That from Proviral Templates. J. Virol. 2009, 83, 8662–8673. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Marsh, J.W. Early Transcription from Nonintegrated DNA in Human Immunodeficiency Virus Infection. J. Virol. 2003, 77, 10376–10382. [Google Scholar] [CrossRef]
- Wu, Y.; Marsh, J.W. Selective Transcription and Modulation of Resting T Cell Activity by Preintegrated HIV DNA. Science 2001, 293, 1503–1506. [Google Scholar] [CrossRef]
- Wu, Y.; Marsh, J.W. Gene Transcription in HIV Infection. Microbes Infect. 2003, 5, 1023–1027. [Google Scholar] [CrossRef]
- Kelly, J.; Beddall, M.H.; Yu, D.; Iyer, S.R.; Marsh, J.W.; Wu, Y. Human Macrophages Support Persistent Transcription from Unintegrated HIV-1 DNA. Virology 2008, 372, 300–312. [Google Scholar] [CrossRef]
- Cara, A.; Cereseto, A.; Lori, F.; Reitz, M.S. HIV-1 Protein Expression from Synthetic Circles of DNA Mimicking the Extrachromosomal Forms of Viral DNA. J. Biol. Chem. 1996, 271, 5393–5397. [Google Scholar] [CrossRef]
- Brussel, A.; Sonigo, P. Evidence for Gene Expression by Unintegrated Human Immunodeficiency Virus Type 1 DNA Species. J. Virol. 2004, 78, 11263–11271. [Google Scholar] [CrossRef]
- Gillim-Ross, L.; Cara, A.; Klotman, M.E. Nef Expressed from Human Immunodeficiency Virus Type 1 Extrachromosomal DNA Downregulates CD4 on Primary CD4+ T Lymphocytes: Implications for Integrase Inhibitors. J. Gen. Virol. 2005, 86, 765–771. [Google Scholar] [CrossRef]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple Effects of Mutations in Human Immunodeficiency Virus Type 1 Integrase on Viral Replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef]
- Levin, A.; Hayouka, Z.; Friedler, A.; Brack-Werner, R.; Volsky, D.J.; Loyter, A. A Novel Role for the Viral Rev Protein in Promoting Resistance to Superinfection by Human Immunodeficiency Virus Type 1. J. Gen. Virol. 2010, 91, 1503–1513. [Google Scholar] [CrossRef]
- Gelderblom, H.C.; Vatakis, D.N.; Burke, S.A.; Lawrie, S.D.; Bristol, G.C.; Levy, D.N. Viral Complementation Allows HIV-1 Replication without Integration. Retrovirology 2008, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Trinité, B.; Ohlson, E.C.; Voznesensky, I.; Rana, S.P.; Chan, C.N.; Mahajan, S.; Alster, J.; Burke, S.A.; Wodarz, D.; Levy, D.N. An HIV-1 Replication Pathway Utilizing Reverse Transcription Products That Fail To Integrate. J. Virol. 2013, 87, 12701–12720. [Google Scholar] [CrossRef]
- Chan, C.N.; Trinité, B.; Lee, C.S.; Mahajan, S.; Anand, A.; Wodarz, D.; Sabbaj, S.; Bansal, A.; Goepfert, P.A.; Levy, D.N. HIV-1 Latency and Virus Production from Unintegrated Genomes Following Direct Infection of Resting CD4 T Cells. Retrovirology 2016, 13, 1. [Google Scholar] [CrossRef]
- Darcis, G.; Berkhout, B. Human Immunodeficiency Virus Resistance to Dolutegravir: Are We Looking in the Wrong Place? J. Infect. Dis. 2018, 218, 2020. [Google Scholar] [CrossRef] [PubMed]
- Dicker, I.B.; Samanta, H.K.; Li, Z.; Hong, Y.; Tian, Y.; Banville, J.; Remillard, R.R.; Walker, M.A.; Langley, D.R.; Krystal, M. Changes to the HIV Long Terminal Repeat and to HIV Integrase Differentially Impact HIV Integrase Assembly, Activity, and the Binding of Strand Transfer Inhibitors. J. Biol. Chem. 2007, 282, 31186–31196. [Google Scholar] [CrossRef]
- Malet, I.; Subra, F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.-G.; Delelis, O. Mutations Located Outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. mBio 2017, 8, e00922-17. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Le Grice, S.F.J. “Binding, Bending and Bonding”: Polypurine Tract-Primed Initiation of plus-Strand DNA Synthesis in Human Immunodeficiency Virus. Int. J. Biochem. Cell Biol. 2004, 36, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Subra, F.; Malet, I.; Leh, H.; Charpentier, C.; Corona, A.; Collin, G.; Descamps, D.; Deprez, E.; Parissi, V.; et al. Mutations in the 3’-PPT Lead to HIV-1 Replication without Integration. J. Virol. 2022, 96, e0067622. [Google Scholar] [CrossRef] [PubMed]
- Kantor, B.; Bayer, M.; Ma, H.; Samulski, J.; Li, C.; McCown, T.; Kafri, T. Notable Reduction in Illegitimate Integration Mediated by a PPT-Deleted, Nonintegrating Lentiviral Vector. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Malet, I.; Subra, F.; Richetta, C.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.-G.; Delelis, O. Reply to Das and Berkhout, “How Polypurine Tract Changes in the HIV-1 RNA Genome Can Cause Resistance against the Integrase Inhibitor Dolutegravir”. mBio 2018, 9, e00623-18. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, A.; Kubota, M.; Shigemi, U.; Ode, H.; Yokomaku, Y.; Kirby, K.A.; Sarafianos, S.G.; Iwatani, Y. Specific Mutations in the HIV-1 G-Tract of the 3’-Polypurine Tract Cause Resistance to Integrase Strand Transfer Inhibitors. J. Antimicrob. Chemother. 2022, 77, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Ferris, A.; Zhao, X.; Pauly, G.; Schneider, J.P.; Burke, T.R.; Hughes, S.H. INSTIs and NNRTIs Potently Inhibit HIV-1 Polypurine Tract Mutants in a Single Round Infection Assay. Viruses 2021, 13, 2501. [Google Scholar] [CrossRef]
- Wei, Y.; Sluis-Cremer, N. Mutations in the HIV-1 3’-Polypurine Tract and Integrase Strand Transfer Inhibitor Resistance. Antimicrob. Agents Chemother. 2021, 65, e02432-20. [Google Scholar] [CrossRef]
- Wijting, I.; Rokx, C.; Boucher, C.; van Kampen, J.; Pas, S.; de Vries-Sluijs, T.; Schurink, C.; Bax, H.; Derksen, M.; Andrinopoulou, E.-R.; et al. Dolutegravir as Maintenance Monotherapy for HIV (DOMONO): A Phase 2, Randomised Non-Inferiority Trial. Lancet HIV 2017, 4, e547–e554. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INSTIs | Dissociative T1/2 (h) | Reference |
|---|---|---|
| RAL | 8.8 | [45] |
| EVG | 2.7 | [45] |
| DTG | 71 | [45] |
| BIC | 163 | [46] |
| INSTIs | Dissociative T1/2 (h) | Possible Integration Events Involving uDNA (2-LTR Circles or Linear Viral DNA) | Emergence of Resistant Strains with Mutations in the IN Gene | Emergence of Resistant Strains without Mutations in the IN gene |
|---|---|---|---|---|
| RAL | 8.8 [45] | ++ | + | − |
| EVG | 2.7 [45] | ++ | + | − |
| DTG | 71 [45] | +/− | +/− | + |
| BIC | 163 [46] | +/− | +/− | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richetta, C.; Tu, N.Q.; Delelis, O. Different Pathways Conferring Integrase Strand-Transfer Inhibitors Resistance. Viruses 2022, 14, 2591. https://doi.org/10.3390/v14122591
Richetta C, Tu NQ, Delelis O. Different Pathways Conferring Integrase Strand-Transfer Inhibitors Resistance. Viruses. 2022; 14(12):2591. https://doi.org/10.3390/v14122591
Chicago/Turabian StyleRichetta, Clémence, Nhat Quang Tu, and Olivier Delelis. 2022. "Different Pathways Conferring Integrase Strand-Transfer Inhibitors Resistance" Viruses 14, no. 12: 2591. https://doi.org/10.3390/v14122591
APA StyleRichetta, C., Tu, N. Q., & Delelis, O. (2022). Different Pathways Conferring Integrase Strand-Transfer Inhibitors Resistance. Viruses, 14(12), 2591. https://doi.org/10.3390/v14122591