
Hepatitis C Core Protein Induces a Genotype-Specific Susceptibility of Hepatocytes to TNF-Induced Death In Vitro and In Vivo

 ,
,  ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
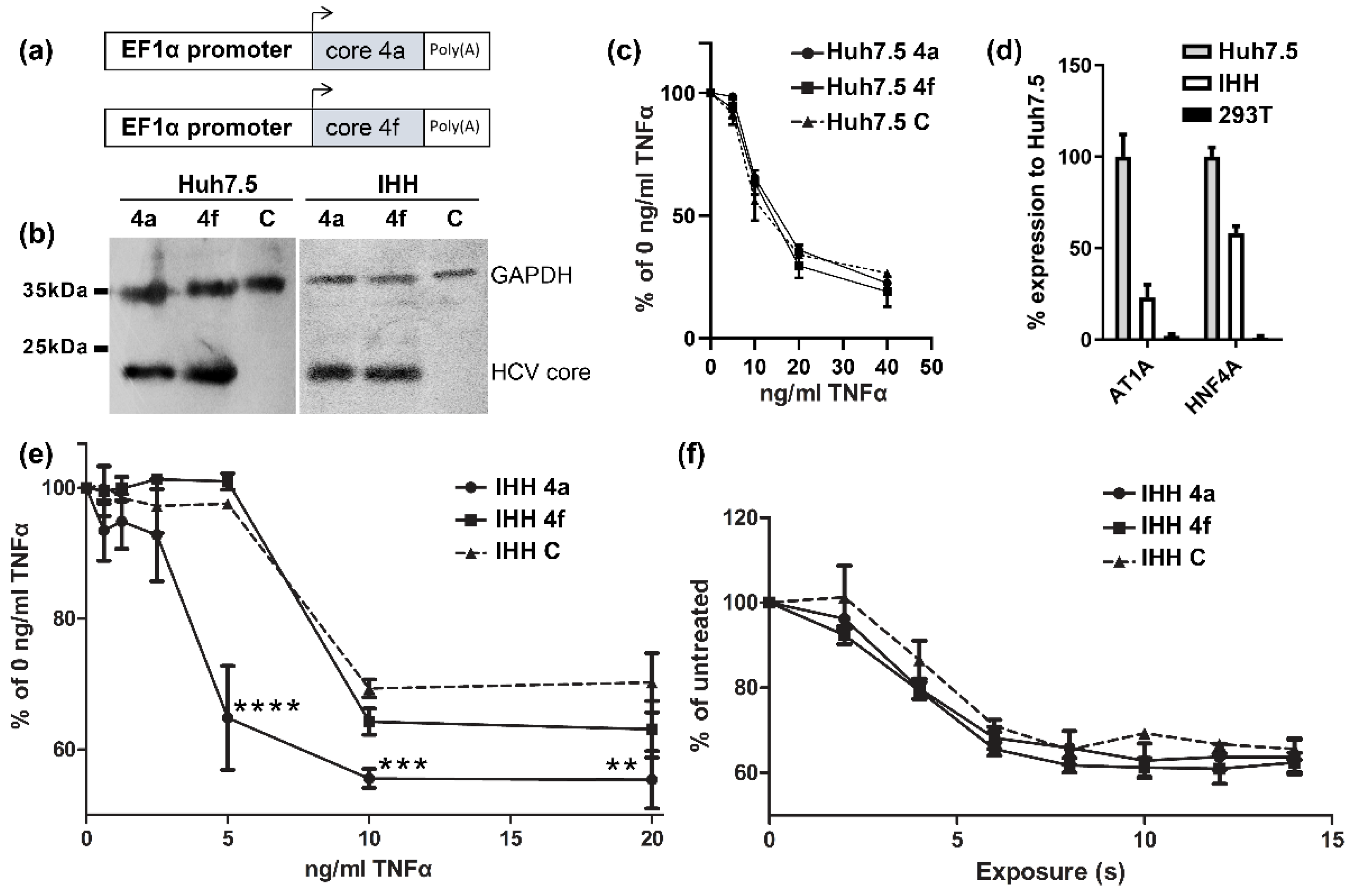
2.1. Cell Lines
2.2. Lentivirus Expression Vectors and Stable Cell Lines
2.3. TNF-Cycloheximide Cytotoxicity Assay
2.4. UV Irradiation Assays
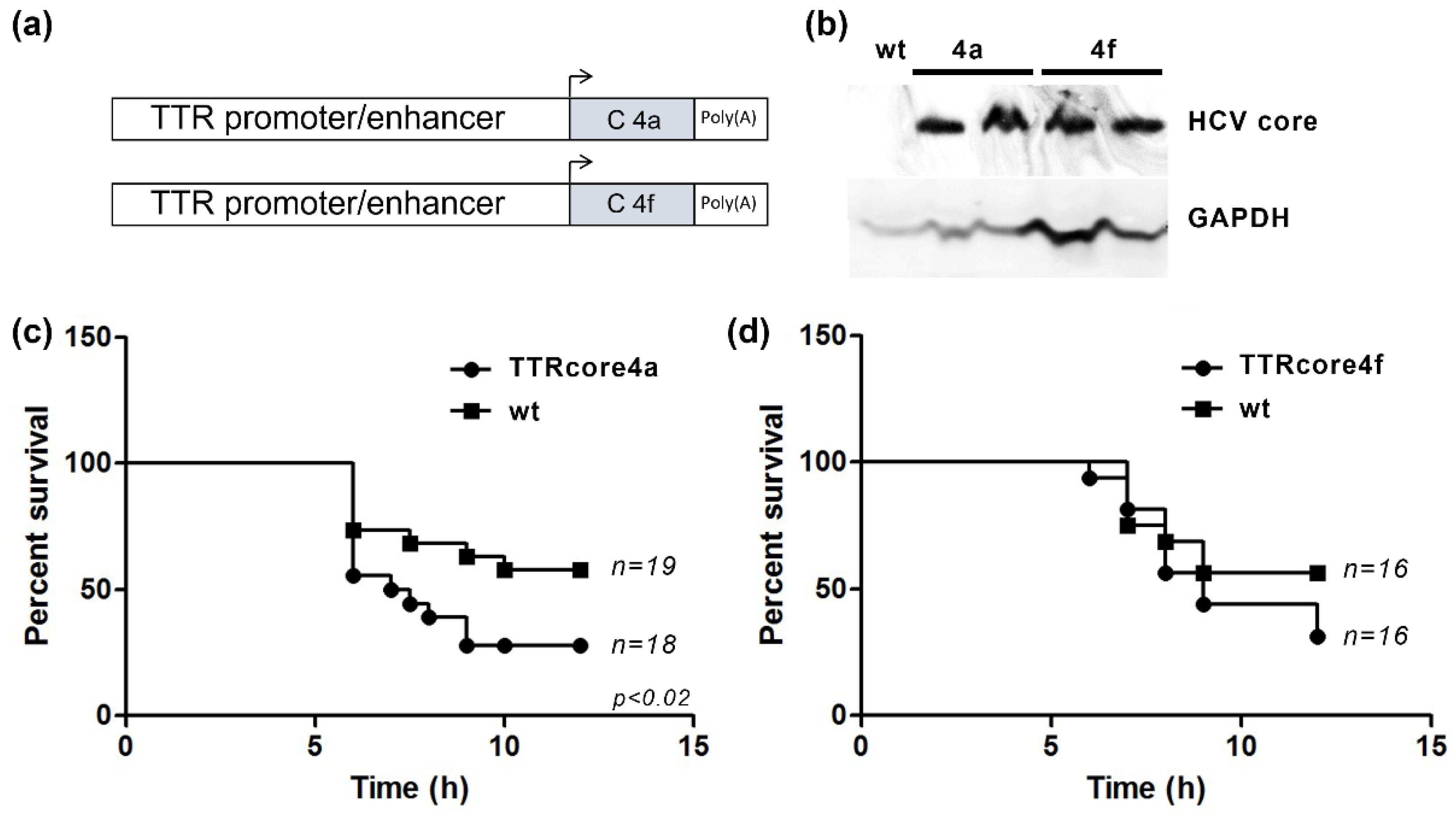
2.5. Transgenic Mice
2.6. Murine Model of Acute Liver Failure
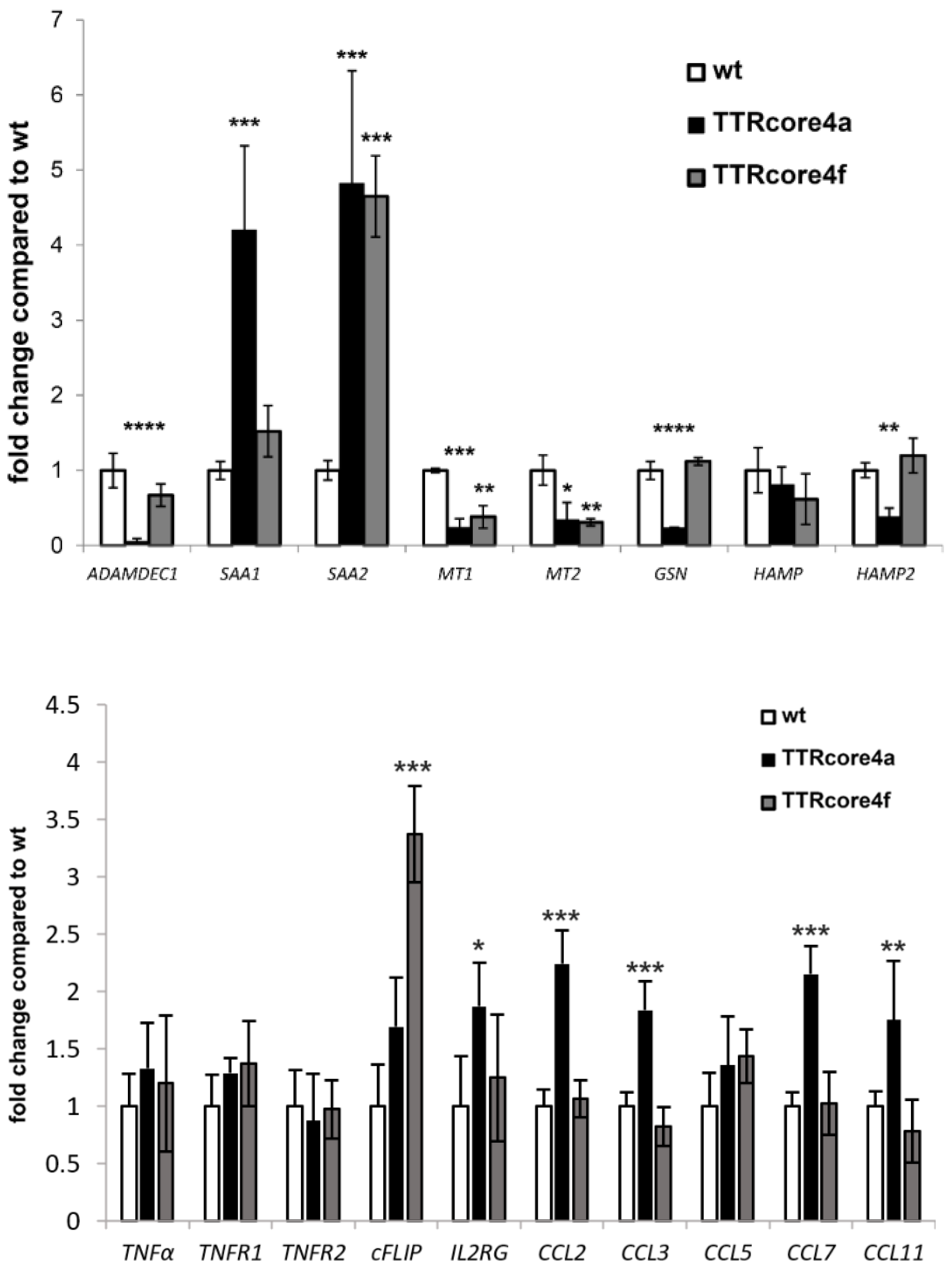
2.7. RNA Sequencing and Bioinformatics Analysis
2.8. Real-Time RT-PCR
3. Results
3.1. HCV Core Presents Differential Susseptibility to TNFα-Induced Death In Vitro
3.2. HCV Core Expression Results in Differential Susseptibility to the LPS/Dgal Hepatic Failure Model
3.3. Subtype-Specific Modulation of Molecular Pathways by HCV Core In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Cheung, M.C.M.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.H.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef]
- Backus, L.I.; Belperio, P.S.; Shahoumian, T.A.; Mole, L.A. Direct-acting antiviral sustained virologic response: Impact on mortality in patients without advanced liver disease. Hepatology 2018, 68, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Jühling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Liang, T.J.; Baumert, T.F. Hepatitis C virus entry: Molecular biology and clinical implications. Hepatology 2006, 44, 527–535. [Google Scholar] [CrossRef]
- Nielsen, S.U.; Bassendine, M.F.; Martin, C.; Lowther, D.; Purcell, P.J.; King, B.J.; Neely, D.; Toms, G.L. Characterization of hepatitis C RNA-containing particles from human liver by density and size. J. Gen. Virol. 2008, 89, 2507–2517. [Google Scholar] [CrossRef]
- Chisari, F.V. Unscrambling hepatitis C virus-host interactions. Nature 2005, 436, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Lin, W.; Kim, S.S.; Yeung, E.; Kamegaya, Y.; Blackard, J.T.; Kim, K.A.; Holtzman, M.J.; Chung, R.T. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J. Virol. 2006, 80, 9226–9235. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yoshida, I.; Takamatsu, M.; Ishido, S.; Fujita, T.; Oka, K.; Hotta, H. Complex formation between hepatitis C virus core protein and p21Waf1/Cip1/Sdi1. Biochem. Biophys. Res. Commun. 2000, 273, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, I.; Oka, K.; Hidajat, R.; Nagano-Fujii, M.; Ishido, S.; Hotta, H. Inhibition of p21/Waf1/Cip1/Sdi1 expression by hepatitis C virus core protein. Microbiol. Immunol. 2001, 45, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Gustot, T. Management of bacterial infections in cirrhosis. J. Hepatol. 2012, 56 (Suppl. 1), S1–S12. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Kasprzak, A.; Zabel, M.; Biczysko, W.; Wysocki, J.; Adamek, A.; Spachacz, R.; Surdyk-Zasada, J. Expression of cytokines (TNF-alpha, IL-1alpha, and IL-2) in chronic hepatitis C: Comparative hybridocytochemical and immunocytochemical study in children and adult patients. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2004, 52, 29–38. [Google Scholar] [CrossRef]
- Manco, M.; Marcellini, M.; Giannone, G.; Nobili, V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am. J. Clin. Pathol. 2007, 127, 954–960. [Google Scholar] [CrossRef]
- Castello, G.; Costantini, S.; Scala, S. Targeting the inflammation in HCV-associated hepatocellular carcinoma: A role in the prevention and treatment. J. Transl. Med. 2010, 8, 109. [Google Scholar] [CrossRef][Green Version]
- Chou, A.H.; Tsai, H.F.; Wu, Y.Y.; Hu, C.Y.; Hwang, L.H.; Hsu, P.I.; Hsu, P.N. Hepatitis C virus core protein modulates TRAIL-mediated apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis signaling pathway. J. Immunol. 2005, 174, 2160–2166. [Google Scholar] [CrossRef]
- Park, J.; Kang, W.; Ryu, S.W.; Kim, W.I.; Chang, D.Y.; Lee, D.H.; Park, D.Y.; Choi, Y.H.; Choi, K.; Shin, E.C.; et al. Hepatitis C virus infection enhances TNFα-induced cell death via suppression of NF-κB. Hepatology 2012, 56, 831–840. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, N.; Taniguchi, H.; Yoshida, H.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein inhibits apoptosis via enhanced Bcl-xL expression. Virology 2002, 296, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Khoshnan, A.; Schneider, R.; Matsumoto, M.; Dennert, G.; Ware, C.; Lai, M.M. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J. Virol. 1998, 72, 3691–3697. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Ware, C.F.; Lai, M.M. Hepatitis C virus core protein enhances FADD-mediated apoptosis and suppresses TRADD signaling of tumor necrosis factor receptor. Virology 2001, 283, 178–187. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ray, R.B.; Meyer, K.; Steele, R.; Shrivastava, A.; Aggarwal, B.B.; Ray, R. Inhibition of tumor necrosis factor (TNF-alpha)-mediated apoptosis by hepatitis C virus core protein. J. Biol. Chem. 1998, 273, 2256–2259. [Google Scholar] [CrossRef]
- Jahan, S.; Khaliq, S.; Siddiqi, M.H.; Ijaz, B.; Ahmad, W.; Ashfaq, U.A.; Hassan, S. Anti-apoptotic effect of HCV core gene of genotype 3a in Huh-7 cell line. Virol. J. 2011, 8, 522. [Google Scholar] [CrossRef]
- Saito, K.; Meyer, K.; Warner, R.; Basu, A.; Ray, R.B.; Ray, R. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 2006, 80, 4372–4379. [Google Scholar] [CrossRef]
- Chan, A.; Patel, K.; Naggie, S. Genotype 3 Infection: The Last Stand of Hepatitis C Virus. Drugs 2017, 77, 131–144. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Hepatitis C virus genetic variability: Pathogenic and clinical implications. Clin. Liver Dis. 2003, 7, 45–66. [Google Scholar] [CrossRef]
- Khaliq, S.; Jahan, S.; Pervaiz, A. Sequence variability of HCV Core region: Important predictors of HCV induced pathogenesis and viral production. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2011, 11, 543–556. [Google Scholar] [CrossRef]
- Backes, P.; Quinkert, D.; Reiss, S.; Binder, M.; Zayas, M.; Rescher, U.; Gerke, V.; Bartenschlager, R.; Lohmann, V. Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 2010, 84, 5775–5789. [Google Scholar] [CrossRef]
- Moustafa, S.; Karakasiliotis, I.; Mavromara, P. Hepatitis C Virus core+1/ARF Protein Modulates the Cyclin D1/pRb Pathway and Promotes Carcinogenesis. J. Virol. 2018, 92, e02036-17. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.P.; Kyrmizi, I.; Cardot, P.; Gonzalez, F.J.; Talianidis, I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol. Cell. Biol. 2010, 30, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Mignon, A.; Rouquet, N.; Fabre, M.; Martin, S.; Pages, J.C.; Dhainaut, J.F.; Kahn, A.; Briand, P.; Joulin, V. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am. J. Respir. Crit. Care Med. 1999, 159, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Carlson, M. R Package, version 3.2.3; org. Mm. eg. db: Genome Wide Annotation for Mouse; Bioconductor: London, UK, 2019. [Google Scholar]
- Carlson, M.; Falcon, S.; Pages, H.; Li, N. R Package; org. Hs. eg. db: Genome Wide Annotation for Human; Bioconductor: London, UK, 2019. [Google Scholar]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.; Wu, G.; Matthews, L. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, D428–D432. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Turkes, E.; Ostendorf, B.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. Bioconductor Version: Release (3.12). 2021. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 12 April 2022).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Aicher, S.; Kakkanas, A.; Cohen, L.; Blumen, B.; Oprisan, G.; Njouom, R.; Meurs, E.F. Differential regulation of the Wnt/β-catenin pathway by hepatitis C virus recombinants expressing core from various genotypes. Sci. Rep. 2018, 8, 11185. [Google Scholar] [CrossRef]
- Ji, Y.R.; Kim, H.J.; Bae, K.B.; Lee, S.; Kim, M.O.; Ryoo, Z.Y. Hepatic serum amyloid A1 aggravates T cell-mediated hepatitis by inducing chemokines via Toll-like receptor 2 in mice. J. Biol. Chem. 2015, 290, 12804–12811. [Google Scholar] [CrossRef] [PubMed]
- Lledó, G.M.; Carrasco, I.; Benítez-Gutiérrez, L.M.; Arias, A.; Royuela, A.; Requena, S.; Cuervas-Mons, V.; de Mendoza, C. Regression of liver fibrosis after curing chronic hepatitis C with oral antivirals in patients with and without HIV coinfection. AIDS 2018, 32, 2347–2352. [Google Scholar] [CrossRef] [PubMed]
- Agwa, R.H.; Elgazzar, M.H.; El-Zayyadi, I.A.; Saed, A.M.; Ghannam, M.A.; Saleh, A. Effect of sustained virological response after direct-acting antivirals on liver fibrosis in patients with chronic HCV infection. Egypt. J. Intern. Med. 2022, 34, 1–8. [Google Scholar] [CrossRef]
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249. [Google Scholar] [CrossRef]
- Dirchwolf, M.; Ruf, A.E. Role of systemic inflammation in cirrhosis: From pathogenesis to prognosis. World J. Hepatol. 2015, 7, 1974–1981. [Google Scholar] [CrossRef]
- Diao, P.; Jia, F.; Wang, X.; Hu, X.; Kimura, T.; Nakajima, T.; Aoyama, T.; Moriya, K.; Koike, K.; Tanaka, N. Mechanisms of steatosis-derived hepatocarcinogenesis: Lessons from HCV core gene transgenic mice. Engineering 2021, 7, 1797–1805. [Google Scholar] [CrossRef]
- Bartosch, B.; Thimme, R.; Blum, H.E.; Zoulim, F. Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 2009, 51, 810–820. [Google Scholar] [CrossRef]
- Mahmoudvand, S.; Shokri, S.; Taherkhani, R.; Farshadpour, F. Hepatitis C virus core protein modulates several signaling pathways involved in hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 42–58. [Google Scholar] [CrossRef]
- Chung, H.; Watanabe, T.; Kudo, M.; Chiba, T. Hepatitis C virus core protein induces homotolerance and cross-tolerance to Toll-like receptor ligands by activation of Toll-like receptor 2. J. Infect. Dis. 2010, 202, 853–861. [Google Scholar] [CrossRef]
- Coenen, M.; Nischalke, H.D.; Krämer, B.; Langhans, B.; Glässner, A.; Schulte, D.; Körner, C.; Sauerbruch, T.; Nattermann, J.; Spengler, U. Hepatitis C virus core protein induces fibrogenic actions of hepatic stellate cells via toll-like receptor 2. Lab. Investig. J. Tech. Methods Pathol. 2011, 91, 1375–1382. [Google Scholar] [CrossRef]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78 Pt 7, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Investig. 2008, 118, 683–694. [Google Scholar] [CrossRef]
- Tiegs, G.; Horst, A.K. TNF in the liver: Targeting a central player in inflammation. Semin. Immunopathol. 2022, 44, 445–459. [Google Scholar] [CrossRef]
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat. Metab. 2021, 3, 1302–1312. [Google Scholar] [CrossRef]
- Yang, Y.M.; Seki, E. TNFalpha in liver fibrosis. Curr. Pathobiol. Rep. 2015, 3, 253–261. [Google Scholar] [CrossRef]
- Jang, J.; Jung, Y.; Chae, S.; Chung, S.I.; Kim, S.M.; Yoon, Y. WNT/β-catenin pathway modulates the TNF-α-induced inflammatory response in bronchial epithelial cells. Biochem. Biophys. Res. Commun. 2017, 484, 442–449. [Google Scholar] [CrossRef]
- Coskun, M.; Olsen, A.K.; Bzorek, M.; Holck, S.; Engel, U.H.; Nielsen, O.H.; Troelsen, J.T. Involvement of CDX2 in the cross talk between TNF-α and Wnt signaling pathway in the colon cancer cell line Caco-2. Carcinogenesis 2014, 35, 1185–1192. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Heyd, F.; Hegyi, K.; Sethi, J.K. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 2007, 14, 1361–1373. [Google Scholar] [CrossRef]
- Hiyama, A.; Yokoyama, K.; Nukaga, T.; Sakai, D.; Mochida, J. A complex interaction between Wnt signaling and TNF-α in nucleus pulposus cells. Arthritis Res. Ther. 2013, 15, R189. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Bradrick, S.; Qiang, G.; Mostafavi, A.; Chaturvedi, G.; Weinman, S.A.; Diehl, A.M.; Jhaveri, R. Up-regulation of Hedgehog pathway is associated with cellular permissiveness for hepatitis C virus replication. Hepatology 2011, 54, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, M.; Yamaji, T.; Saito, K.; Shirasago, Y.; Satomura, K.; Endo, T.; Fukasawa, M.; Hanada, K.; Osada, N. Identification of Characteristic Genomic Markers in Human Hepatoma HuH-7 and Huh7.5.1-8 Cell Lines. Front. Genet. 2020, 11, 546106. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Gaines, G.C.; Rosenberg, J.; Minter, R.; Bahjat, F.R.; Rectenwald, J.; MacKay, S.L.; Edwards, C.K., 3rd; Moldawer, L.L. LPS-induced liver injury in D-galactosamine-sensitized mice requires secreted TNF-alpha and the TNF-p55 receptor. American journal of physiology. Regul. Integr. Comp. Physiol. 2000, 278, R1202–R1209. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Hope, G.; Boulant, S.; McLauchlan, J. Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J. Biol. Chem. 2008, 283, 16850–16859. [Google Scholar] [CrossRef]
- Kumagai, T.; Fan, S.; Smith, A.M. ADAMDEC1 and its role in inflammatory disease and cancer. Met. Med. 2020, 7, 15–28. [Google Scholar] [CrossRef]
- O’Shea, N.R.; Chew, T.S.; Dunne, J.; Marnane, R.; Nedjat-Shokouhi, B.; Smith, P.J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Critical Role of the Disintegrin Metalloprotease ADAM-like Decysin-1 [ADAMDEC1] for Intestinal Immunity and Inflammation. J. Crohn’s Colitis 2016, 10, 1417–1427. [Google Scholar] [CrossRef]
- Leifeld, L.; Fink, K.; Debska, G.; Fielenbach, M.; Schmitz, V.; Sauerbruch, T.; Spengler, U. Anti-apoptotic function of gelsolin in fas antibody-induced liver failure in vivo. Am. J. Pathol. 2006, 168, 778–785. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Li, D.; Xie, P.; Zhao, S.; Zhao, J.; Yao, Y.; Zhao, Y.; Ren, G.; Liu, X. Hepatocytes derived increased SAA1 promotes intrahepatic platelet aggregation and aggravates liver inflammation in NAFLD. Biochem. Biophys. Res. Commun. 2021, 555, 54–60. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, D.; Hu, Y.; Li, W.; Liu, F.; Zhu, X.; Li, X.; Zhang, H.; Bai, H.; Yang, Q.; et al. Serum amyloid A1 exacerbates hepatic steatosis via TLR4-mediated NF-κB signaling pathway. Mol. Metab. 2022, 59, 101462. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W.; Zuo, R.; Liu, C.; Shu, Q.; Ying, H.; Sun, K. Induction of pro-inflammatory genes by serum amyloid A1 in human amnion fibroblasts. Sci. Rep. 2017, 7, 693. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; Parnell, G.; Booth, D.; Douglas, M.W.; George, J.; Ahlenstiel, G. The antiviral role of zinc and metallothioneins in hepatitis C infection. J. Viral Hepat. 2018, 25, 491–501. [Google Scholar] [CrossRef]
- Hino, K.; Harada, M. Metal metabolism and liver. Liver Syst. Dis. 2016, 123–146. [Google Scholar] [CrossRef]
- Muendlein, H.I.; Jetton, D.; Connolly, W.M.; Eidell, K.P.; Magri, Z.; Smirnova, I.; Poltorak, A. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 2020, 367, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Panayotova-Dimitrova, D.; Feoktistova, M.; Ploesser, M.; Kellert, B.; Hupe, M.; Horn, S.; Makarov, R.; Jensen, F.; Porubsky, S.; Schmieder, A.; et al. cFLIP regulates skin homeostasis and protects against TNF-induced keratinocyte apoptosis. Cell Rep. 2013, 5, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Burm, R.; Collignon, L.; Mesalam, A.A.; Meuleman, P. Animal Models to Study Hepatitis C Virus Infection. Front. Immunol. 2018, 9, 1032. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | p-Value | FDR | Fold Change |
|---|---|---|---|
| Upregulated Genes | |||
| HHLA1 | 0.0000000 | 0.0000000 | 53.00 |
| TNNC2 | 0.0000000 | 0.0000091 | 21.67 |
| ACTA1 | 0.0000056 | 0.0045909 | 11.00 |
| SERPINE1 | 0.0000416 | 0.0195843 | 9.33 |
| SAA1 | 0.0000000 | 0.0000441 | 5.96 |
| SAA2 | 0.0000000 | 0.0000762 | 5.82 |
| GM42674 | 0.0000206 | 0.0107402 | 4.96 |
| S100A4 | 0.0000206 | 0.0107402 | 4.96 |
| SELENBP2 | 0.0000013 | 0.0012631 | 3.79 |
| MUP17 | 0.0000011 | 0.0011299 | 3.58 |
| MUP7 | 0.0000077 | 0.0059425 | 3.55 |
| MUP15 | 0.0000297 | 0.0144767 | 3.39 |
| CYP7B1 | 0.0000017 | 0.0015311 | 3.12 |
| HSP90AA1 | 0.0000236 | 0.0119081 | 2.96 |
| MUP11 | 0.0001180 | 0.0462593 | 2.91 |
| TSKU | 0.0000117 | 0.0075599 | 2.90 |
| BAG3 | 0.0000853 | 0.0366297 | 2.61 |
| Downregulated Genes | |||
| ADAMDEC1 | 0.0000000 | 0.0000000 | −23.32 |
| CLEC3B | 0.0000000 | 0.0000079 | −15.50 |
| INO80DOS | 0.0000651 | 0.0296855 | −13.13 |
| FBLN1 | 0.0000003 | 0.0003058 | −8.59 |
| CRISPLD2 | 0.0000000 | 0.0000348 | −7.64 |
| HTRA3 | 0.0000133 | 0.0080787 | −6.45 |
| CYP26A1 | 0.0000007 | 0.0007748 | −6.07 |
| MATN2 | 0.0001204 | 0.0462593 | −6.00 |
| LPAR1 | 0.0000098 | 0.0071805 | −5.91 |
| ELN | 0.0001141 | 0.0462593 | −5.18 |
| GSN | 0.0000000 | 0.0000011 | −5.18 |
| NIPAL1 | 0.0000199 | 0.0107402 | −4.64 |
| HAMP2 | 0.0000000 | 0.0000004 | −4.57 |
| DPT | 0.0000119 | 0.0075599 | −4.56 |
| COL6A3 | 0.0000205 | 0.0107402 | −3.58 |
| MGP | 0.0000057 | 0.0045909 | −3.44 |
| MT2 | 0.0000001 | 0.0001359 | −3.20 |
| AQP8 | 0.0000001 | 0.0001509 | −3.14 |
| CYP4A14 | 0.0000719 | 0.0318185 | −2.96 |
| COL1A1 | 0.0000922 | 0.0384517 | −2.78 |
| MT1 | 0.0000104 | 0.0072522 | −2.52 |
| 8430408G22RIK | 0.0001333 | 0.0499041 | −2.06 |
| Gene Symbol | p-Value | FDR | Fold Change |
|---|---|---|---|
| Upregulated Genes | |||
| IKBIP | 0.0000023 | 0.0033358 | 8.10 |
| SAA2 | 0.0000003 | 0.0005697 | 5.85 |
| HAMP | 0.0000612 | 0.0470078 | 3.29 |
| Downregulated Genes | |||
| LOX | 0.0000000 | 0.0000010 | −21.19 |
| ANKRD1 | 0.0000165 | 0.0185512 | −14.10 |
| DMBT1 | 0.0000000 | 0.0000000 | −12.44 |
| S100A4 | 0.0000000 | 0.0000000 | −10.19 |
| GM42674 | 0.0000000 | 0.0000000 | −10.05 |
| RP24-361O20.1 | 0.0000005 | 0.0008336 | −9.67 |
| GM16198 | 0.0000372 | 0.0339678 | −7.80 |
| DDIT4 | 0.0000000 | 0.0000053 | −6.77 |
| S100A6 | 0.0000000 | 0.0000013 | −6.09 |
| GM20649 | 0.0000145 | 0.0176227 | −5.42 |
| MUP-PS17 | 0.0000002 | 0.0003379 | −5.14 |
| MEG3 | 0.0000433 | 0.0351342 | −3.97 |
| DNAAF5 | 0.0000326 | 0.0317683 | −3.95 |
| EIF4G1 | 0.0000040 | 0.0053495 | −3.71 |
| LCOR | 0.0000178 | 0.0185512 | −3.57 |
| CYP26B1 | 0.0000404 | 0.0347101 | −2.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moustafa, S.; Kassela, K.; Bampali, M.; Dovrolis, N.; Kakkanas, A.; Beloukas, A.; Mavromara, P.; Karakasiliotis, I. Hepatitis C Core Protein Induces a Genotype-Specific Susceptibility of Hepatocytes to TNF-Induced Death In Vitro and In Vivo. Viruses 2022, 14, 2521. https://doi.org/10.3390/v14112521
Moustafa S, Kassela K, Bampali M, Dovrolis N, Kakkanas A, Beloukas A, Mavromara P, Karakasiliotis I. Hepatitis C Core Protein Induces a Genotype-Specific Susceptibility of Hepatocytes to TNF-Induced Death In Vitro and In Vivo. Viruses. 2022; 14(11):2521. https://doi.org/10.3390/v14112521
Chicago/Turabian StyleMoustafa, Savvina, Katerina Kassela, Maria Bampali, Nikolas Dovrolis, Athanassios Kakkanas, Apostolos Beloukas, Penelope Mavromara, and Ioannis Karakasiliotis. 2022. "Hepatitis C Core Protein Induces a Genotype-Specific Susceptibility of Hepatocytes to TNF-Induced Death In Vitro and In Vivo" Viruses 14, no. 11: 2521. https://doi.org/10.3390/v14112521
APA StyleMoustafa, S., Kassela, K., Bampali, M., Dovrolis, N., Kakkanas, A., Beloukas, A., Mavromara, P., & Karakasiliotis, I. (2022). Hepatitis C Core Protein Induces a Genotype-Specific Susceptibility of Hepatocytes to TNF-Induced Death In Vitro and In Vivo. Viruses, 14(11), 2521. https://doi.org/10.3390/v14112521