
Healthcare-Associated COVID-19 across Five Pandemic Waves: Prediction Models and Genomic Analyses

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Setting and Timeline
2.2. Definitions and Inclusions
2.3. Objectives and Study Design
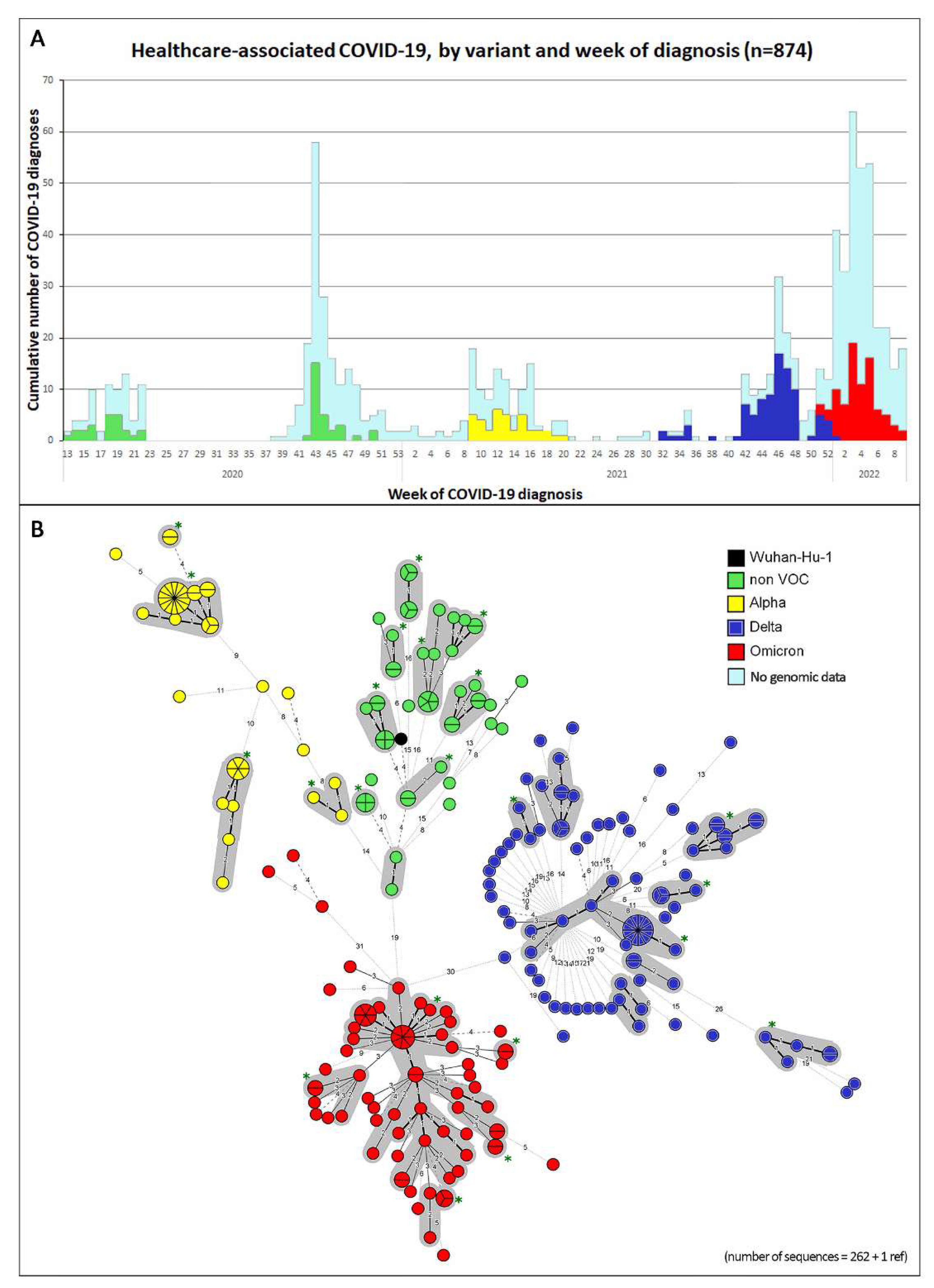
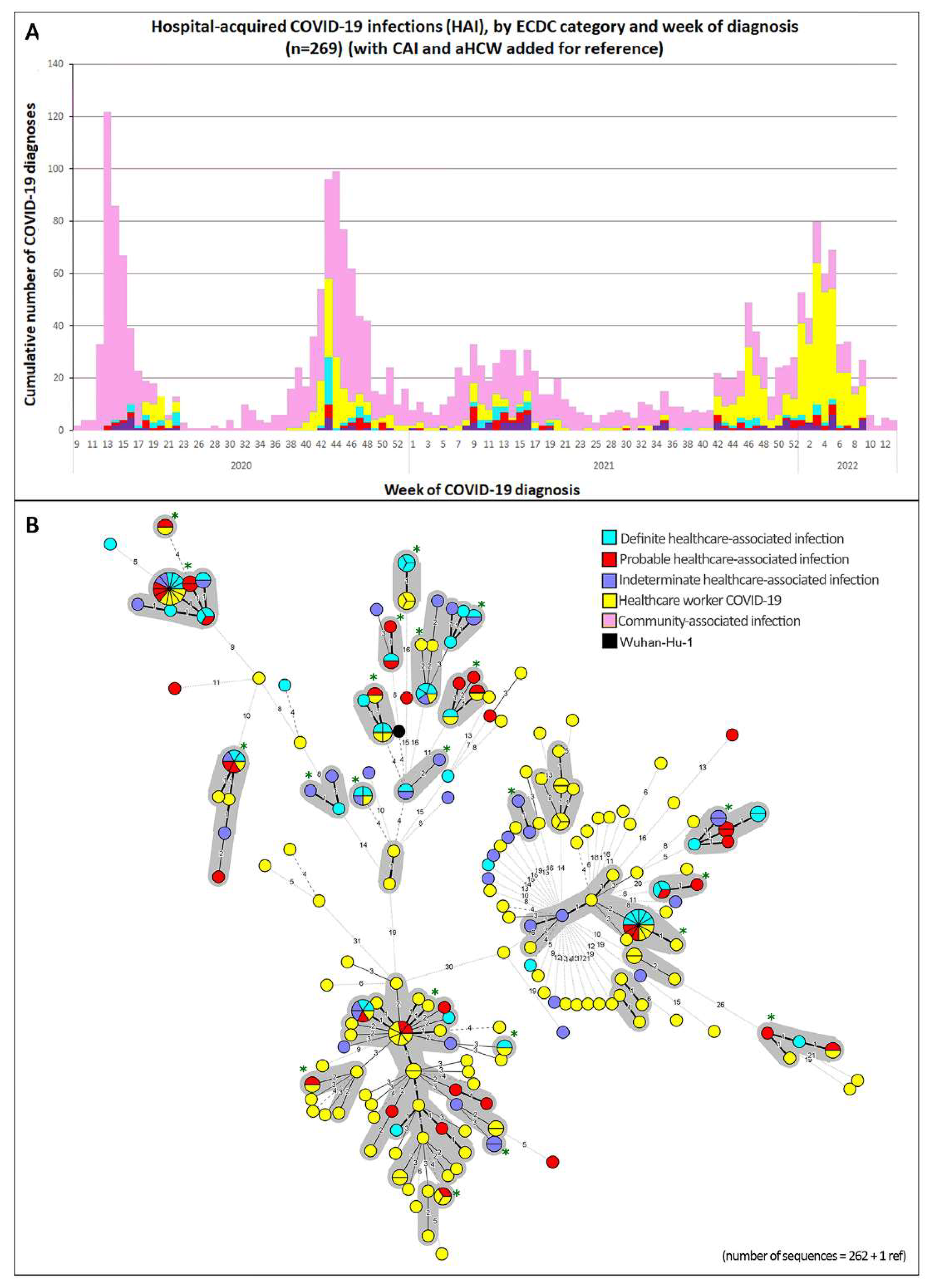
2.4. Laboratory: Inclusion of Samples for Genetic Sequencing and Genomic Cluster Analyses
2.5. Statistical and Epidemiological Analyses
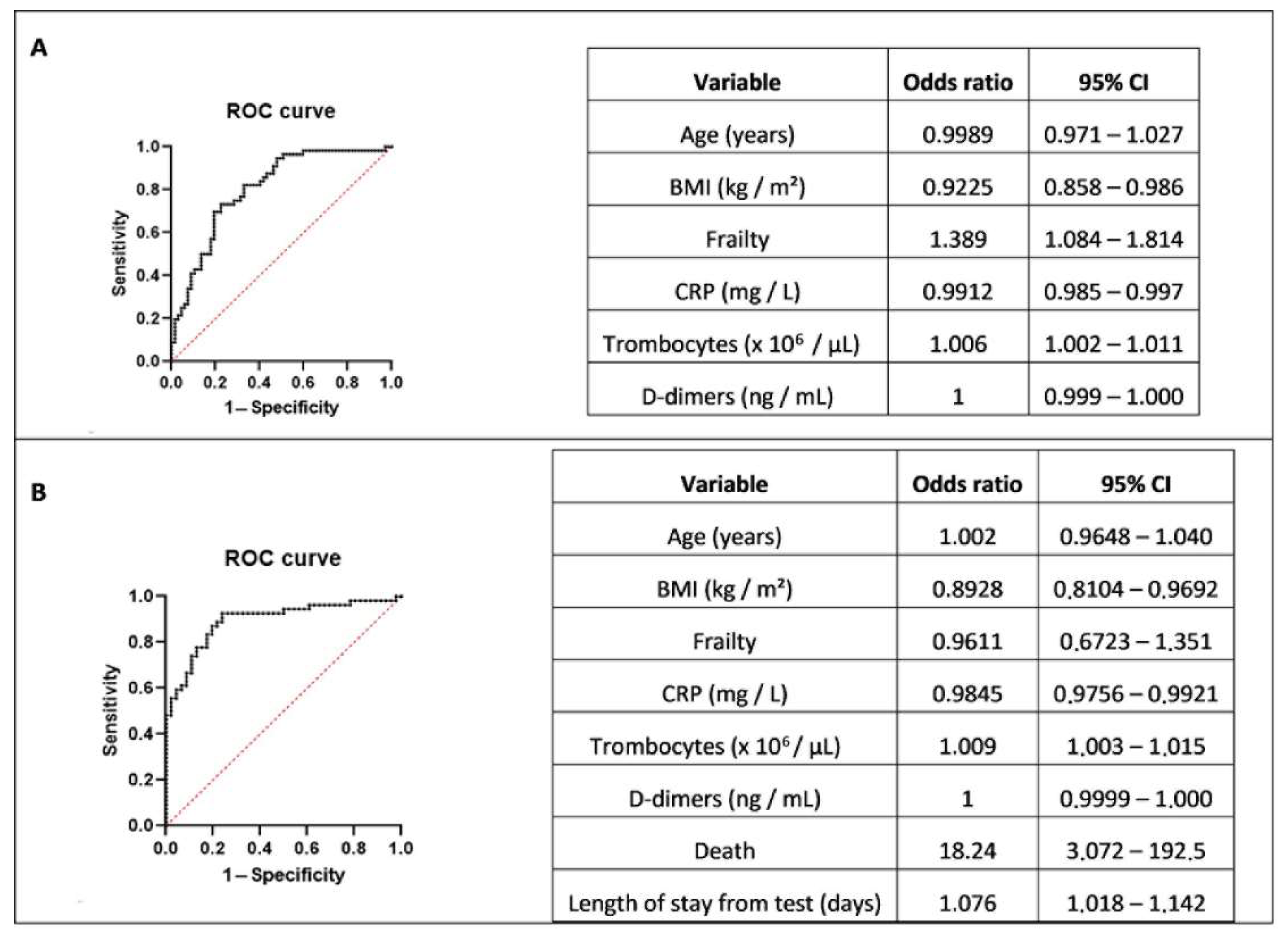
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA—J. Am. Med. Assoc. 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Marago, I.; Minen, I. Hospital-Acquired COVID-19 Infection—The Magnitude of the Problem. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Drew, D.A.; Graham, M.S.; Joshi, A.D.; Guo, C.G.; Ma, W.; Mehta, R.S.; Warner, E.T.; Sikavi, D.R.; Lo, C.H.; et al. Risk of COVID-19 among Front-Line Health-Care Workers and the General Community: A Prospective Cohort Study. Lancet Public Health 2020, 5, e475–e483. [Google Scholar] [CrossRef]
- Rivett, L.; Sridhar, S.; Sparkes, D.; Routledge, M.; Jones, N.K.; Forrest, S.; Young, J.; Pereira-Dias, J.; Hamilton, W.L.; Ferris, M.; et al. Screening of Healthcare Workers for SARS-CoV-2 Highlights the Role of Asymptomatic Carriage in COVID-19 Transmission. Elife 2020, 9, e58728. [Google Scholar] [CrossRef] [PubMed]
- Barranco, R.; du Tremoul, L.V.B.; Ventura, F. Hospital-Acquired Sars-Cov-2 Infections in Patients: Inevitable Conditions or Medical Malpractice? Int. J. Environ. Res. Public Health 2021, 18, 489. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, S.; Mishra, S.; Gandy, A.; Unwin, H.J.T.; Mellan, T.A.; Coupland, H.; Whittaker, C.; Zhu, H.; Berah, T.; Eaton, J.W.; et al. Estimating the Effects of Non-Pharmaceutical Interventions on COVID-19 in Europe. Nature 2020, 584, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic Characteristics and Clinical Effect of the Emergent SARS-CoV-2 B.1.1.7 Lineage in London, UK: A Whole-Genome Sequencing and Hospital-Based Cohort Study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R.; et al. Community Transmission and Viral Load Kinetics of the SARS-CoV-2 Delta (B.1.617.2) Variant in Vaccinated and Unvaccinated Individuals in the UK: A Prospective, Longitudinal, Cohort Study. Lancet Infect. Dis. 2021, 22, 183–195. [Google Scholar] [CrossRef]
- Demeester, S.; Demuyser, T.; Fauconnier, C.; Heestermans, R.; Orlando, C.; Depreter, B.; Jochmans, K. Routine Haematology Parameters in COVID-19 Patients and Clinical Outcome: A Belgian Single-Centre Study. Int. J. Lab. Hematol. 2020, 6, 19–20. [Google Scholar] [CrossRef]
- Khonyongwa, K.; Taori, S.K.; Soares, A.; Desai, N.; Sudhanva, M.; Bernal, W.; Schelenz, S.; Curran, L.A. Incidence and Outcomes of Healthcare-Associated COVID-19 Infections: Significance of Delayed Diagnosis and Correlation with Staff Absence. J. Hosp. Infect. 2020, 106, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Surveillance Definitions for COVID-19. Available online: https://www.ecdc.europa.eu/en/covid-19/surveillance/surveillance-definitions (accessed on 25 July 2021).
- Meredith, L.; Hamilton, W.; Warne, B.; Houldcroft, C.; Hosmillo, M.; Jahun, A.; Curran, M.; Parmar, S.; Caller, L.; Caddy, S.; et al. Rapid Implementation of Real-Time SARS-CoV-2 Sequencing to Investigate Healthcare-Associated COVID-19 Infections. BMJ 2020, 20, 1263–1271. [Google Scholar] [CrossRef]
- Paltansing, S.; Sikkema, R.S.; de Man, S.J.; Koopmans, M.P.G.; Oude Munnink, B.B.; de Man, P. Transmission of SARS-CoV-2 among Healthcare Workers and Patients in a Teaching Hospital in the Netherlands Confirmed by Whole-Genome Sequencing. J. Hosp. Infect. 2021, 110, 178–183. [Google Scholar] [CrossRef] [PubMed]
- SARS-CoV2 Genome Sequencing Protocol (1200 bp Amplicon “Midnight” Primer Set, Using Nanopore Rapid Kit). Available online: https://www.protocols.io/view/sars-cov2-genome-sequencing-protocol-1200bp-amplic-btsrnnd6 (accessed on 13 May 2021).
- Artic Network. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 29 July 2021).
- GISAID—European Union. Available online: https://www.gisaid.org/phylodynamics/european-union/ (accessed on 13 May 2021).
- Severe Acute Respiratory Syndrome Coronavirus 2 Isolate Wuhan-Hu-1, Co-Nucleotide—NCBI. Available online: https://www.ncbi.nlm.nih.gov/nuccore/1798174254 (accessed on 26 July 2021).
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron Variant Result in Stronger Binding to Human ACE2 Receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Case Source COVID-19 | Definition |
|---|---|
| CAI | Symptoms or sample present on admission or with onset on day 1 or 2 after admission (or on days 3–7 with a strong suspicion of community transmission). |
| Indeterminate HAI | Symptom onset or sample present on days 3–7 after admission with insufficient information on the source of infection to assign to another category. |
| Probable HAI | Symptom onset or sample present on days 8–14 after admission (or on days 3–7 and a strong suspicion of healthcare transmission). |
| Definite HAI | Symptom onset or sample present on day > 14 after admission. |
| COVID-19 Wave in Belgium | Date | Hospitalized Patients | aHCW | ||
|---|---|---|---|---|---|
| CAI | HAI | ||||
| Symptomatic (Random Selection for Comparison) | Total (% of CA + HA) | Symptomatic (% of Total HA) | Number | ||
| Wave 1 | 2 March 2020–7 June 2020 | 365 (37) | 43 (10.5) | 36 (83.7) | 29 |
| Between waves 1 and 2 | 8 June 2020–6 September 2020 | 39 (3) | 0 (0.0) | NA | 0 |
| Wave 2 | 7 September 2020–3 January 2021 | 409 (41) | 59 (12.6) | 34 (57.6) | 132 |
| Wave 3 | 4 January 2021–4 July 2021 | 320 (32) | 72 (22.5) | 48 (66.7) | 51 |
| Between waves 3 and 4 | 5 July 2021–31 October 2021 | 151 (15) | 18 (10.7) | 8 (44.4) | 25 |
| Wave 4 | 1 November 2021–26 December 2021 | 158 (16) | 26 (14.1) | 16 (61.5) | 90 |
| Wave 5 | 27 December 2021–6 March 2022 | 154 (15) | 51 (24.9) | 23 (45.1) | 278 |
| Total | 1596 (159) | 269 (14.4) | 165 (61.3) | 605 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demuyser, T.; Seyler, L.; Buttiens, R.; Soetens, O.; Van Nedervelde, E.; Caljon, B.; Praet, J.; Seyler, T.; Boeckmans, J.; Meert, J.; et al. Healthcare-Associated COVID-19 across Five Pandemic Waves: Prediction Models and Genomic Analyses. Viruses 2022, 14, 2292. https://doi.org/10.3390/v14102292
Demuyser T, Seyler L, Buttiens R, Soetens O, Van Nedervelde E, Caljon B, Praet J, Seyler T, Boeckmans J, Meert J, et al. Healthcare-Associated COVID-19 across Five Pandemic Waves: Prediction Models and Genomic Analyses. Viruses. 2022; 14(10):2292. https://doi.org/10.3390/v14102292
Chicago/Turabian StyleDemuyser, Thomas, Lucie Seyler, Rhea Buttiens, Oriane Soetens, Els Van Nedervelde, Ben Caljon, Jessy Praet, Thomas Seyler, Joost Boeckmans, Jessy Meert, and et al. 2022. "Healthcare-Associated COVID-19 across Five Pandemic Waves: Prediction Models and Genomic Analyses" Viruses 14, no. 10: 2292. https://doi.org/10.3390/v14102292
APA StyleDemuyser, T., Seyler, L., Buttiens, R., Soetens, O., Van Nedervelde, E., Caljon, B., Praet, J., Seyler, T., Boeckmans, J., Meert, J., Vanstokstraeten, R., Martini, H., Crombé, F., Piérard, D., Allard, S. D., & Wybo, I. (2022). Healthcare-Associated COVID-19 across Five Pandemic Waves: Prediction Models and Genomic Analyses. Viruses, 14(10), 2292. https://doi.org/10.3390/v14102292