
Molecular Characterization and Cross-Reactivity of Feline Calicivirus Circulating in Southwestern China

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Information and Treatment
2.2. Primer Design
2.3. RNA Extraction and RT-PCR for VP1 Gene
2.4. Virus Isolation and Complete Genome Amplification
2.5. FCV Sequences Analysis
2.6. Preparation of Antigen and Serum Virus Neutralization Assay for FCV Isolates
3. Results
3.1. RT-PCR Survey of Clinical Samples
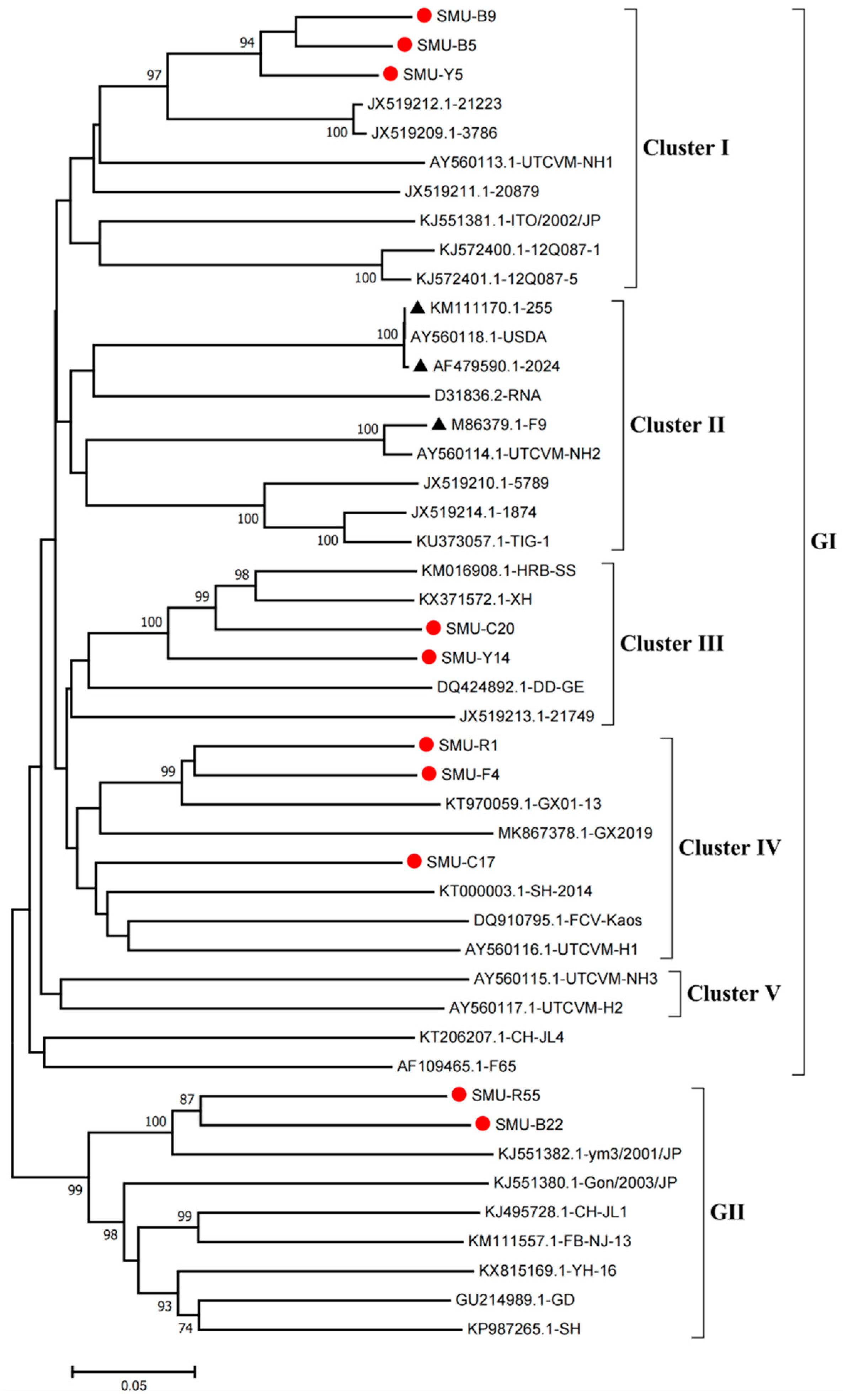
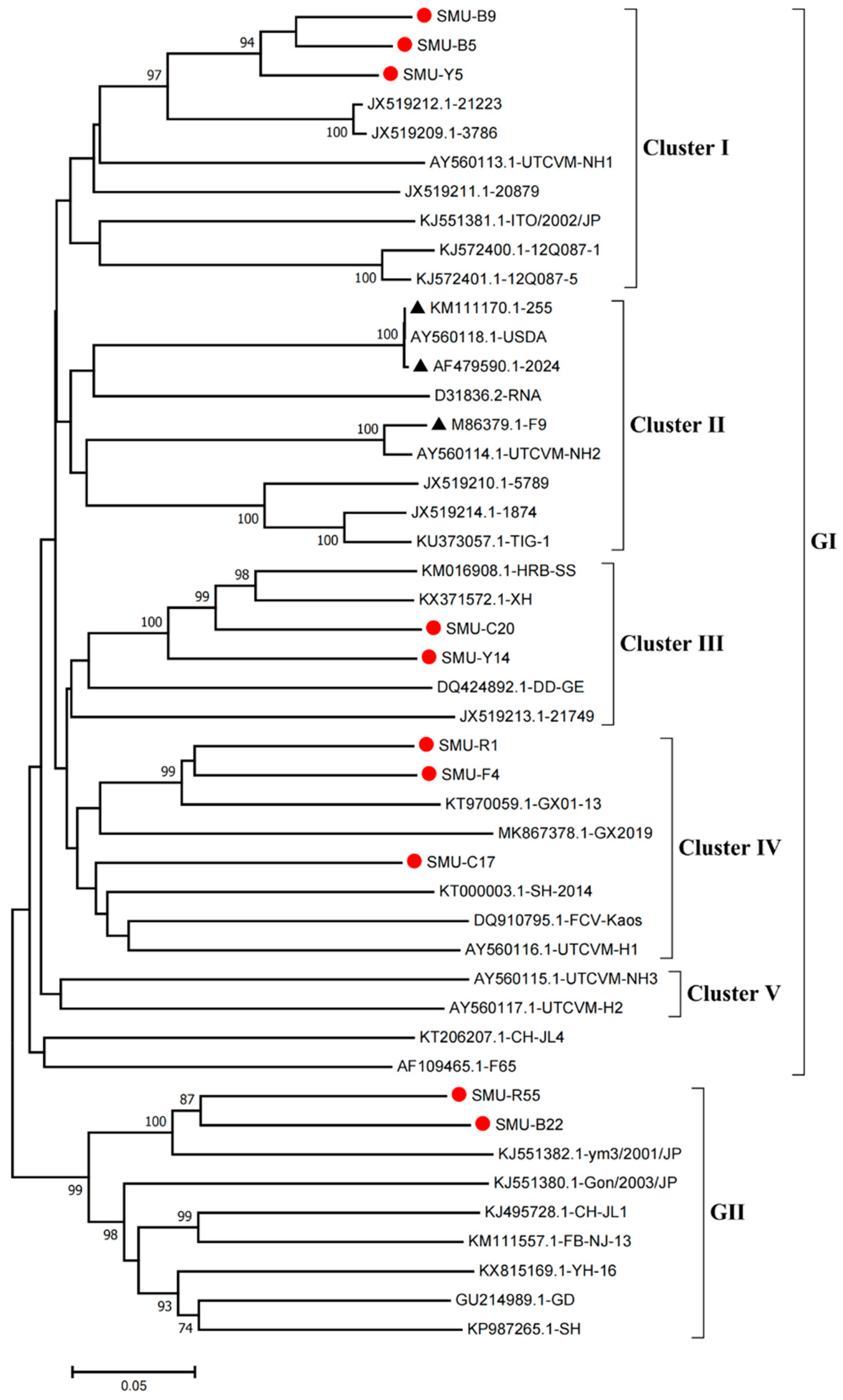
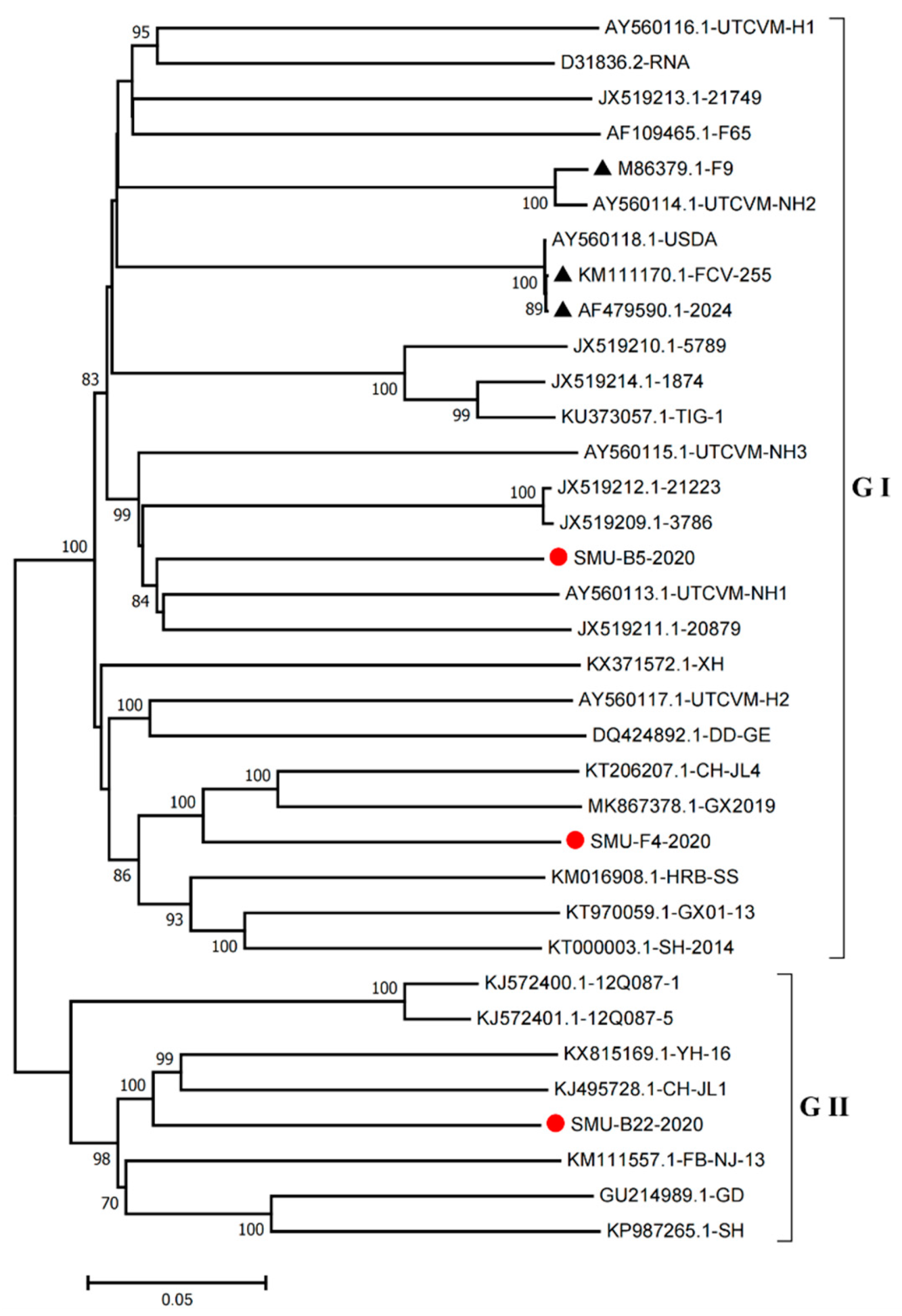
3.2. Sequence Alignment and Phylogenetic Analysis of the VP1 Gene
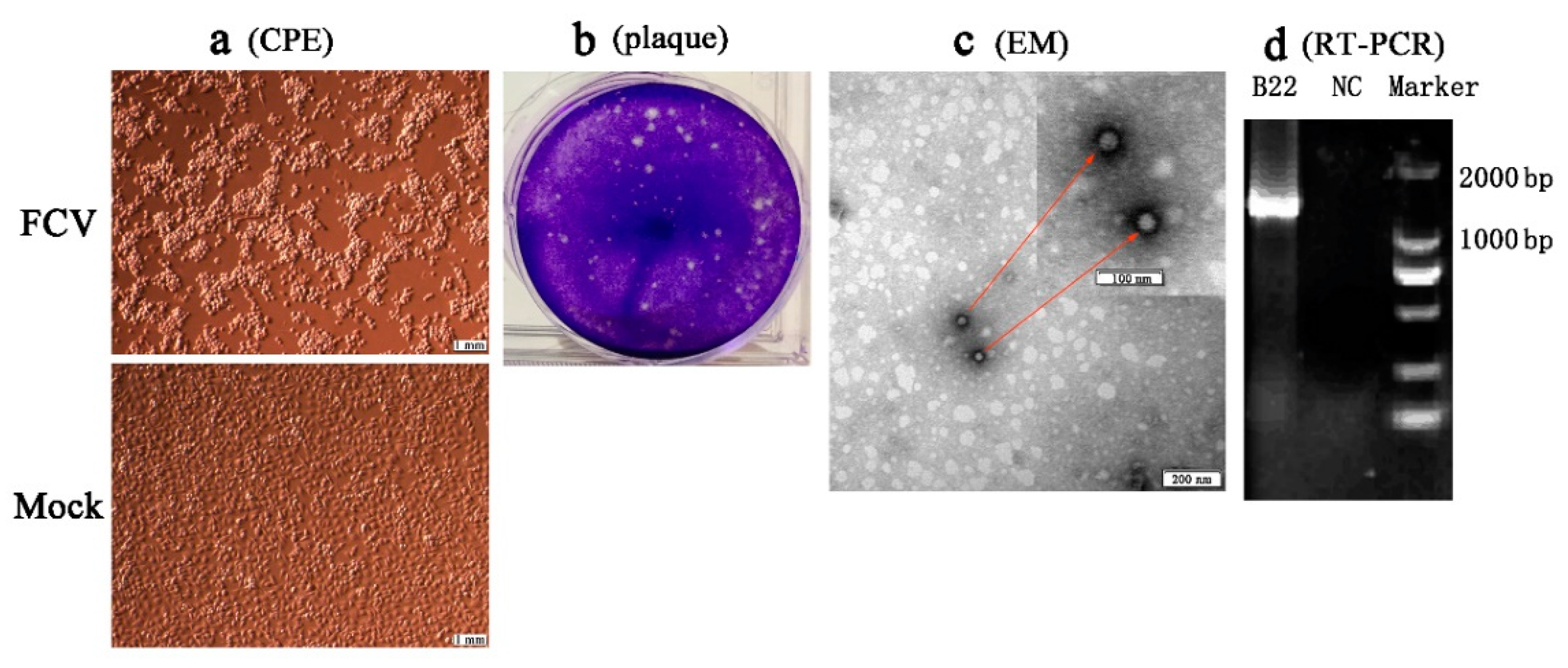
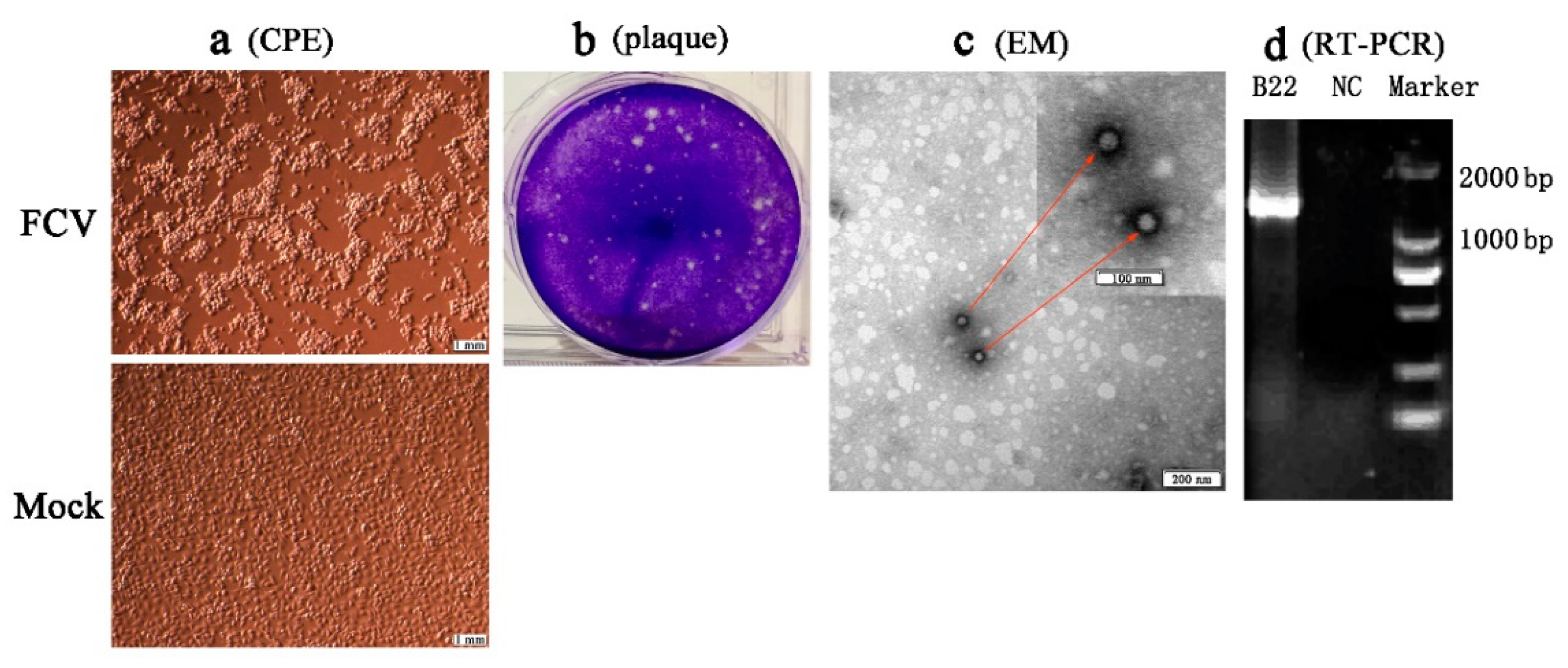
3.3. Isolation and Identification of FCV Strains
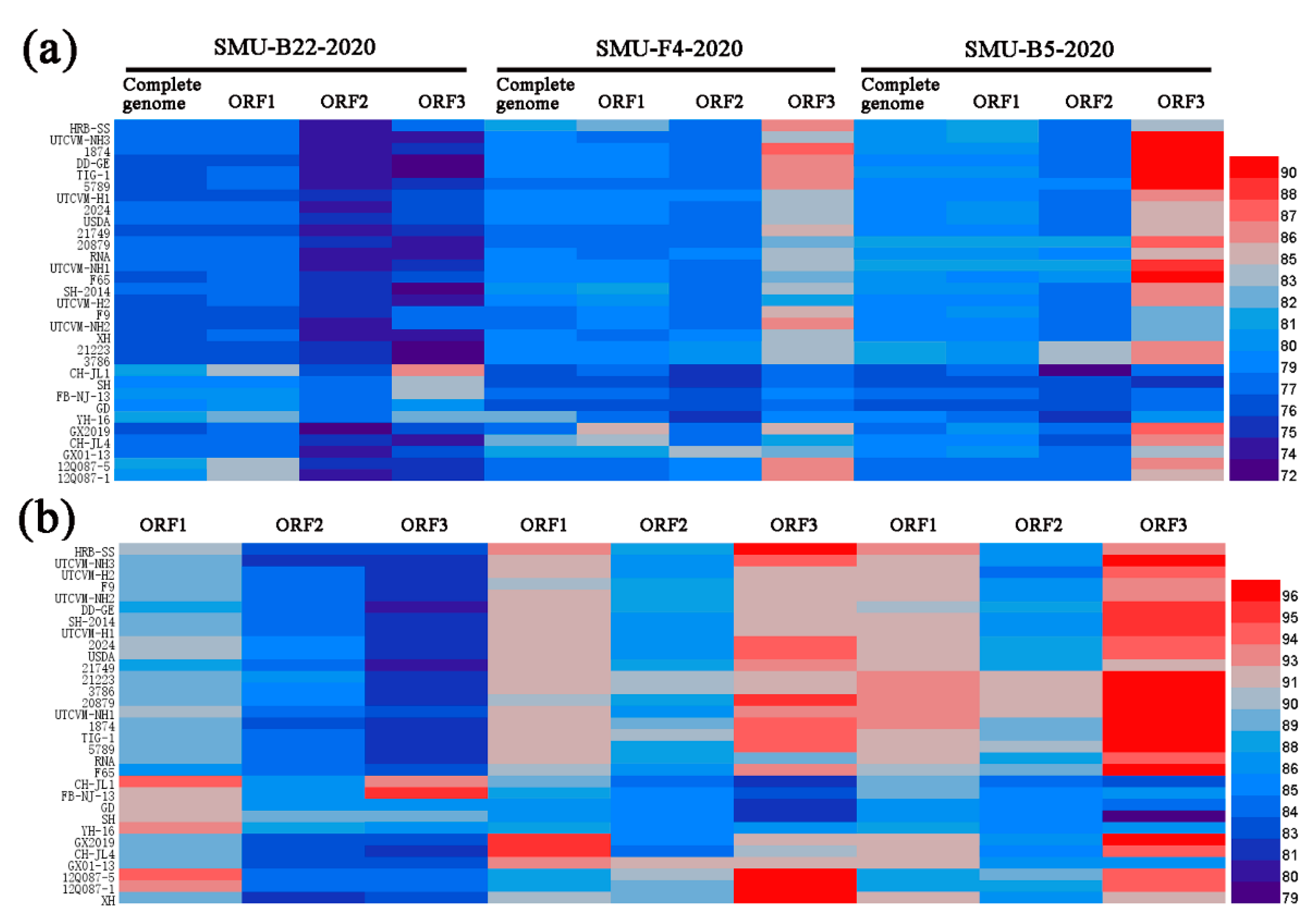
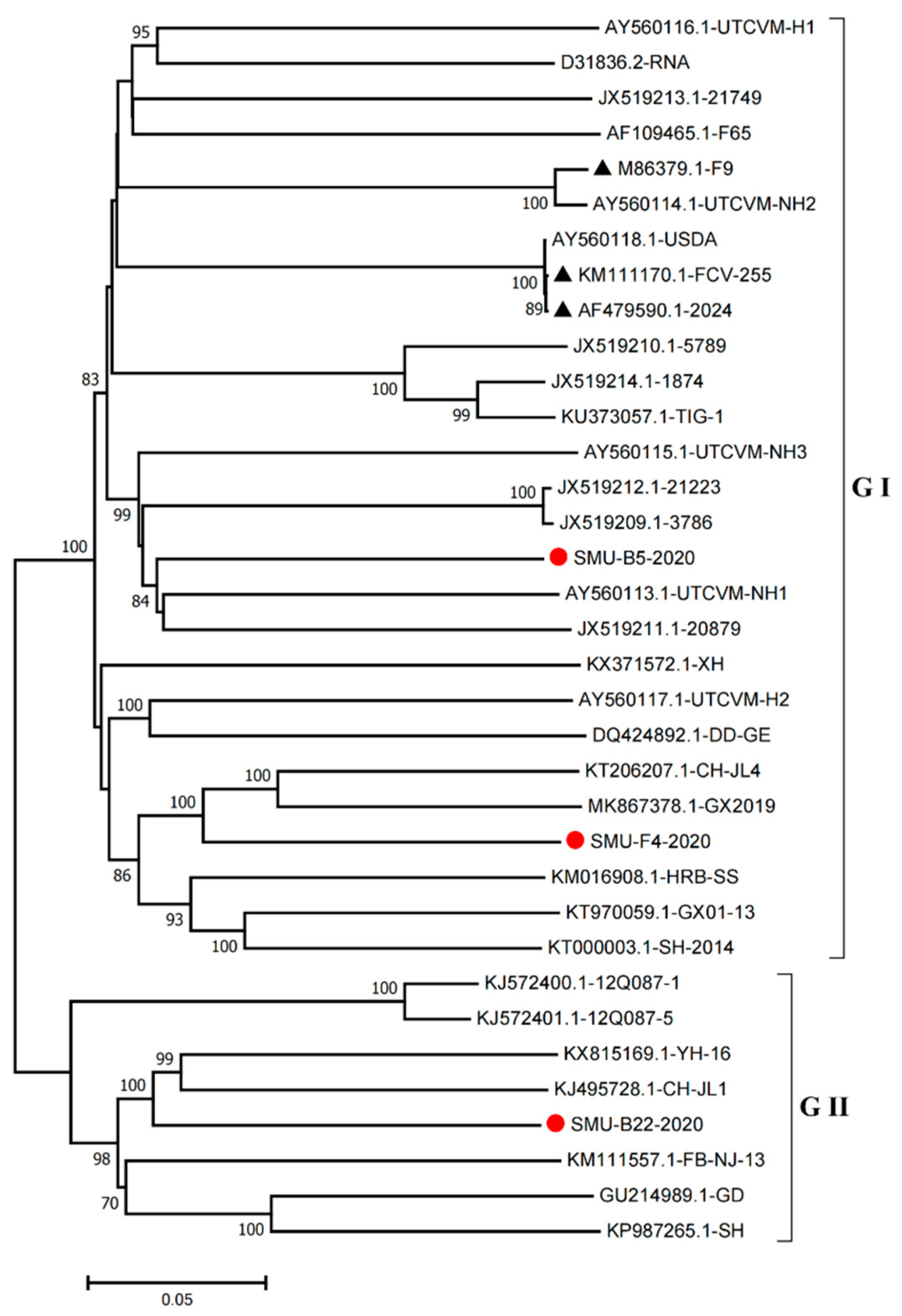
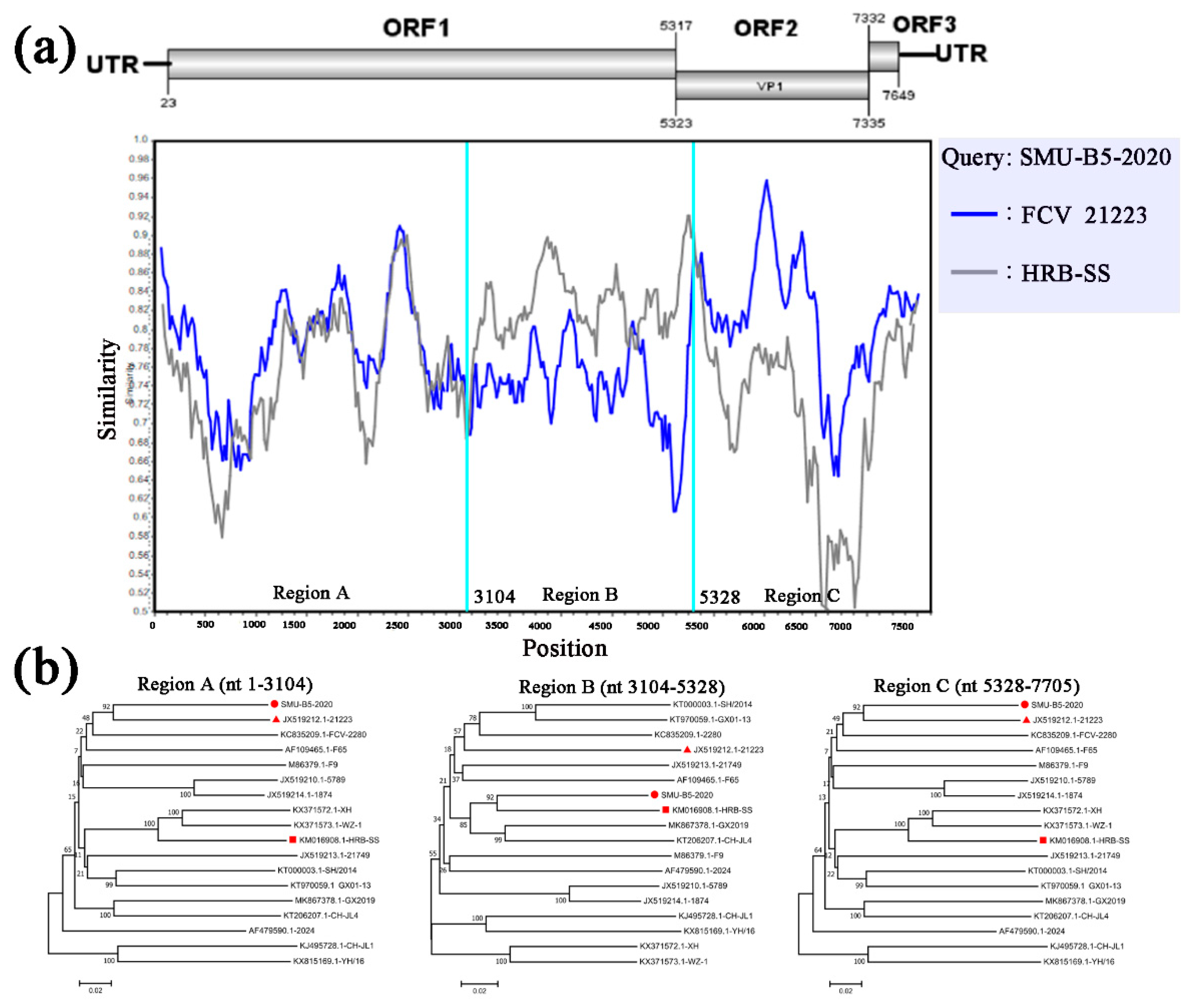
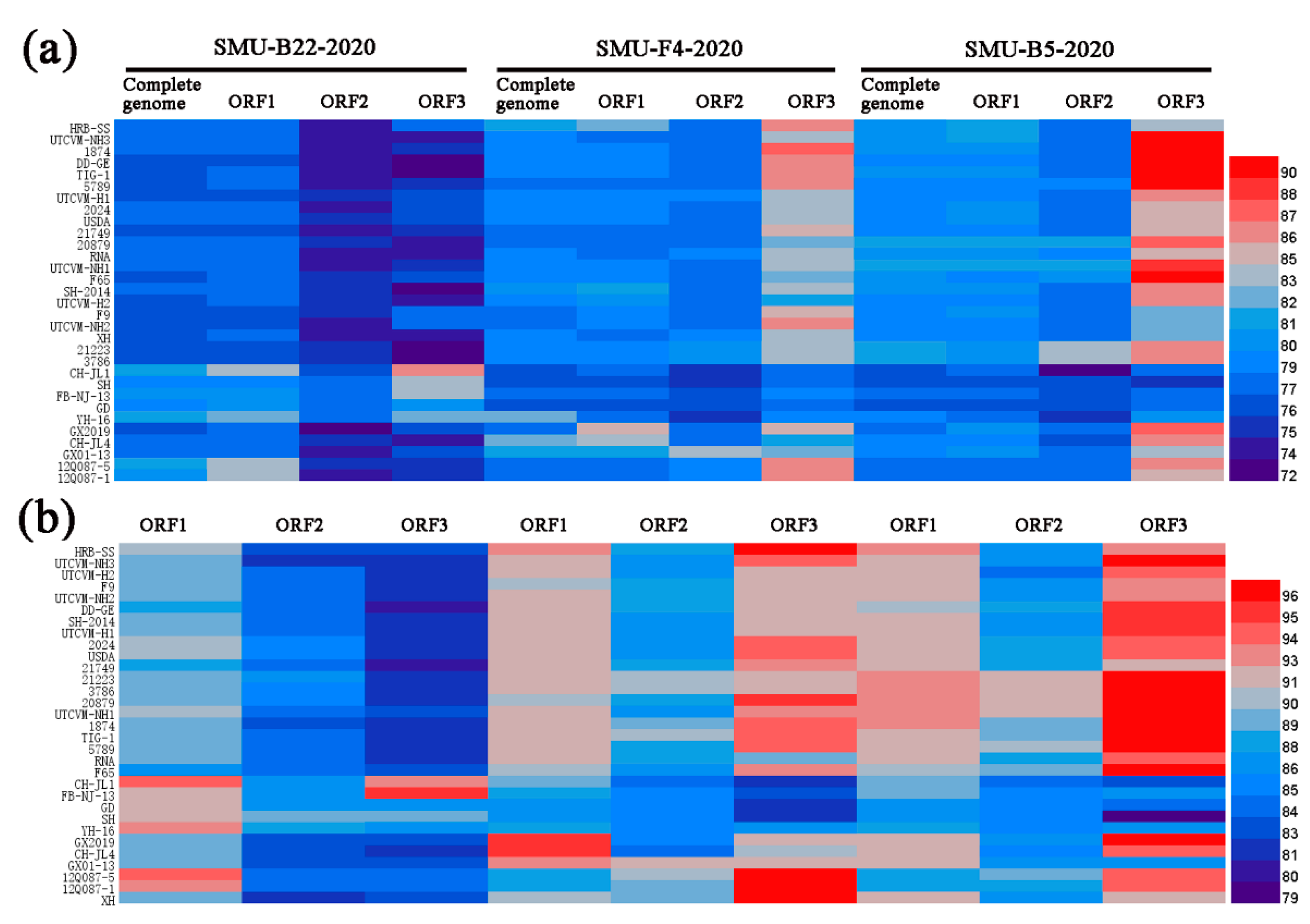
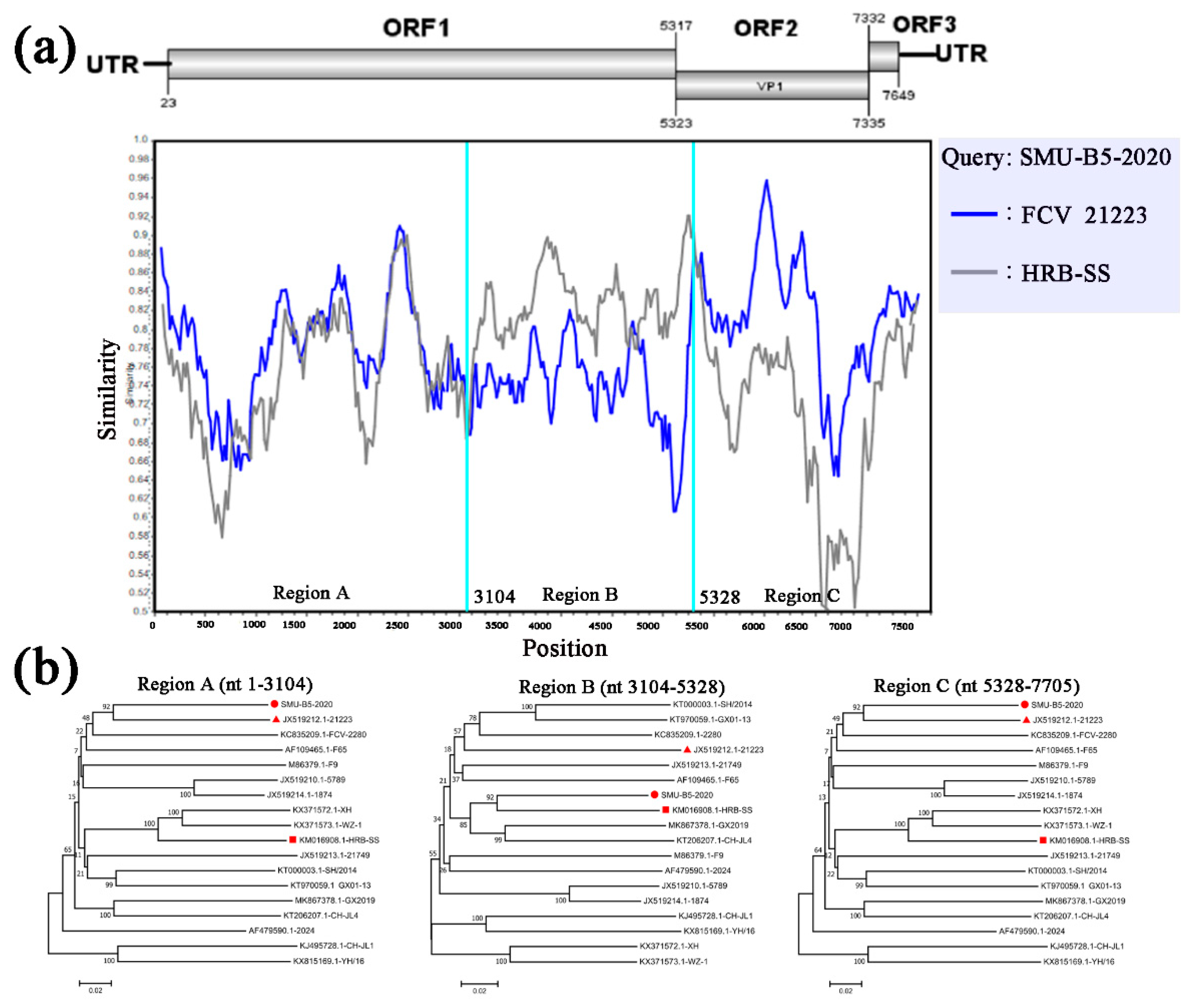
3.4. Full-Length Genome Sequence Analyses of Three New FCV Isolates
3.5. Serum Neutralization Antibody Titers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radford, A.D.; Coyne, K.P.; Dawson, S.; Porter, C.J.; Gaskell, R.M. Feline calicivirus. Vet. Res. 2007, 38, 319–335. [Google Scholar] [CrossRef] [Green Version]
- Fastier, L.B. A new feline virus isolated in tissue culture. Am. J. Vet. Res. 1957, 18, 382–389. [Google Scholar]
- Bannasch, M.J.; Foley, J.E. Epidemiologic evaluation of multiple respiratory pathogens in cats in animal shelters. J. Feline Med. Surg. 2005, 7, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Sanchez-Vizcaino, F.; McGahie, D.; Lesbros, C.; Almeras, T.; Howarth, D.; O’Hara, V.; Dawson, S.; Radford, A.D. European molecular epidemiology and strain diversity of feline calicivirus. Vet. Rec. 2016, 178, 114–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Deng, M.; Peng, Z.; Hu, R.; Chen, H.; Wu, B. Genetic and phylogenetic analysis of feline calicivirus isolates in china. Vet. J. 2017, 220, 24–27. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Elliott, J.B.; Glasgow, A.; Poland, A.; Keel, K. An isolated epizootic of hemorrhagic-like fever in cats caused by a novel and highly virulent strain of feline calicivirus. Vet. Microbiol. 2000, 73, 281–300. [Google Scholar] [CrossRef]
- Radford, A.D.; Sommerville, L.M.; Dawson, S.; Kerins, A.M.; Ryvar, R.; Gaskell, R.M. Molecular analysis of isolates of feline calicivirus from a population of cats in a rescue shelter. Vet. Rec. 2001, 149, 477–481. [Google Scholar] [CrossRef]
- Schorr-Evans, E.M.; Poland, A.; Johnson, W.E.; Pedersen, N.C. An epizootic of highly virulent feline calicivirus disease in a hospital setting in New England. J. Feline Med. Surg. 2003, 5, 217–226. [Google Scholar] [CrossRef]
- Hurley, K.E.; Pesavento, P.A.; Pedersen, N.C.; Poland, A.M.; Wilson, E.; Foley, J.E. An outbreak of virulent systemic feline calicivirus disease. J. Am. Vet. Med. Assoc. 2004, 224, 241–249. [Google Scholar] [CrossRef]
- Reynolds, B.S.; Poulet, H.; Pingret, J.L.; Jas, D.; Brunet, S.; Lemeter, C.; Etievant, M.; Boucraut-Baralon, C. A nosocomial outbreak of feline calicivirus associated virulent systemic disease in france. J. Feline Med. Surg. 2009, 11, 633–644. [Google Scholar] [CrossRef]
- Battilani, M.; Vaccari, F.; Carelle, M.S.; Morandi, F.; Benazzi, C.; Kipar, A.; Dondi, F.; Scagliarini, A. Virulent feline calicivirus disease in a shelter in italy: A case description. Res. Vet. Sci. 2013, 95, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Pesavento, P.A.; Chang, K.O.; Parker, J.S. Molecular virology of feline calicivirus. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; He, W.; Du, F.; Wu, G.; Wu, B.; Zhou, Z. Analysis of the codon usage of the orf2 gene of feline calicivirus. Infect. Genet. Evol. 2017, 54, 54–59. [Google Scholar] [CrossRef]
- Sato, Y.; Ohe, K.; Murakami, M.; Fukuyama, M.; Furuhata, K.; Kishikawa, S.; Suzuki, Y.; Kiuchi, A.; Hara, M.; Ishikawa, Y.; et al. Phylogenetic analysis of field isolates of feline calcivirus (fcv) in japan by sequencing part of its capsid gene. Vet. Res. Commun. 2002, 26, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Henzel, A.; Sa e Silva, M.; Luo, S.; Lovato, L.T.; Weiblen, R. Genetic and phylogenetic analyses of capsid protein gene in feline calicivirus isolates from rio grande do sul in southern brazil. Virus Res. 2012, 163, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, X.; Ying, Y.; Wang, K.; Dong, H.; Gao, C.; Yang, S.; Hu, G. Isolation and phylogenetic analysis of three feline calicivirus strains from domestic cats in jilin province, china. Arch. Virol. 2017, 162, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Caringella, F.; Elia, G.; Decaro, N.; Martella, V.; Lanave, G.; Varello, K.; Catella, C.; Diakoudi, G.; Carelli, G.; Colaianni, M.L.; et al. Feline calicivirus infection in cats with virulent systemic disease, italy. Res. Vet. Sci. 2019, 124, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, B.; Lanave, G.; Di Profio, F.; Melegari, I.; Marsilio, F.; Camero, M.; Catella, C.; Capozza, P.; Banyai, K.; Barrs, V.R.; et al. Identification of feline calicivirus in cats with enteritis. Transbound. Emerg. Dis. 2020, 67, 2579–2588. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, Y.K.; Kim, Y.S.; Na, E.J.; Kim, Y.J.; Oem, J.K. Intergenic recombination in feline calicivirus associated with a hemorrhagic-like disease in the Republic of Korea. Acta Virol. 2021, 65, 232–236. [Google Scholar] [CrossRef]
- Radford, A.D.; Bennett, M.; McArdle, F.; Dawson, S.; Turner, P.C.; Glenn, M.A.; Gaskell, R.M. The use of sequence analysis of a feline calicivirus (fcv) hypervariable region in the epidemiological investigation of fcv related disease and vaccine failures. Vaccine 1997, 15, 1451–1458. [Google Scholar] [CrossRef]
- Afonso, M.M.; Pinchbeck, G.L.; Smith, S.L.; Daly, J.M.; Gaskell, R.M.; Dawson, S.; Radford, A.D. A multi-national european cross-sectional study of feline calicivirus epidemiology, diversity and vaccine cross-reactivity. Vaccine 2017, 35, 2753–2760. [Google Scholar] [CrossRef]
- Smith, S.L.; Afonso, M.M.; Pinchbeck, G.L.; Gaskell, R.M.; Dawson, S.; Radford, A.D. Temporally separated feline calicivirus isolates do not cluster phylogenetically and are similarly neutralised by high-titre vaccine strain fcv-f9 antisera in vitro. J. Feline Med. Surg. 2020, 22, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Marsilio, F.; Di Martino, B.; Decaro, N.; Buonavoglia, C. A novel nested pcr for the diagnosis of calicivirus infections in the cat. Vet. Microbiol. 2005, 105, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Coyne, K.P.; Jones, B.R.; Kipar, A.; Chantrey, J.; Porter, C.J.; Barber, P.J.; Dawson, S.; Gaskell, R.M.; Radford, A.D. Lethal outbreak of disease associated with feline calicivirus infection in cats. Vet. Rec. 2006, 158, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Wardley, R.C.; Gaskell, R.M.; Povey, R.C. Feline respiratory viruses—Their prevalence in clinically healthy cats. J. Small Anim. Pract. 1974, 15, 579–586. [Google Scholar] [CrossRef]
- Coutts, A.J.; Dawson, S.; Willoughby, K.; Gaskell, R.M. Isolation of feline respiratory viruses from clinically healthy cats at UK cat shows. Vet. Rec. 1994, 135, 555–556. [Google Scholar]
- Helps, C.R.; Lait, P.; Damhuis, A.; Bjornehammar, U.; Bolta, D.; Brovida, C.; Chabanne, L.; Egberink, H.; Ferrand, G.; Fontbonne, A.; et al. Factors associated with upper respiratory tract disease caused by feline herpesvirus, feline calicivirus, chlamydophila felis and bordetella bronchiseptica in cats: Experience from 218 european catteries. Vet. Rec. 2005, 156, 669–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Ara, P.; Labriola, C.; Sala, E.; Spada, E.; Magistrelli, S.; Lauzi, S. Prevalence of serum antibody titres against feline panleukopenia, herpesvirus and calicivirus infections in stray cats of milan, italy. Prev. Vet. Med. 2019, 167, 32–38. [Google Scholar] [CrossRef]
- Wang, K.; Pei, Z.; Hu, G. First report of feline calicivirus (fcv) infection in stray cats in northeast china. Pol. J. Vet. Sci. 2017, 20, 595–598. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Liu, D.; Guo, D.; Liu, M.; Li, Y.; Qu, L. Complete genome sequence of feline calicivirus strain hrb-ss from a cat in heilongjiang province, northeastern China. Genome Announc. 2014, 2, e00698-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xin, T.; Wei, J.; Jiang, Y.; Liu, X.; Song, W.; Guo, X.; Yuan, W.; Cui, Y.; Zhu, H.; et al. Isolation and phylogenetic analysis of strains of feline calicivirus in beijing, China. Arch. Virol. 2021, 166, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- Seal, B.S.; Neill, J.D. Capsid protein gene sequence of feline calicivirus isolates 255 and llk: Further evidence for capsid protein configuration among feline caliciviruses. Virus Genes 1995, 9, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, E.; Muylkens, B.; Mauroy, A.; Ziant, D.; Delwiche, T.; Thiry, E. Experimental evidence of recombination in murine noroviruses. J. Gen. Virol. 2010, 91, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Ludwig-Begall, L.F.; Mauroy, A.; Thiry, E. Norovirus recombinants: Recurrent in the field, recalcitrant in the lab—A scoping review of recombination and recombinant types of noroviruses. J. Gen. Virol. 2018, 99, 970–988. [Google Scholar] [CrossRef]
- Coyne, K.P.; Reed, F.C.; Porter, C.J.; Dawson, S.; Gaskell, R.M.; Radford, A.D. Recombination of feline calicivirus within an endemically infected cat colony. J. Gen. Virol. 2006, 87, 921–926. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Collection Site | Year | No. of Samples | No. of Positives | Positive Rate (%) | Location | Clinical Sign |
|---|---|---|---|---|---|---|---|
| 1 | Chengdu | 2017 | 5 | 5 | 100 | Shelter | Conjunctivitis; nasal discharge |
| 2 | Chengdu | 2017 | 5 | 0 | 0 | Shelter | Nasal discharge |
| 3 | Chongqing | 2018 | 8 | 4 | 50 | Shelter | Conjunctivitis |
| 4 | Chengdu | 2018 | 15 | 6 | 40 | Shelter | Conjunctivitis |
| 5 | Chongqing | 2018 | 11 | 3 | 27.3 | Shelter | Conjunctivitis |
| 6 | Chengdu | 2018 | 4 | 2 | 50 | Shelter | Nasal discharge |
| 7 | Chongqing | 2018 | 6 | 2 | 33.3 | Shelter | Nasal discharge |
| 8 | Chengdu | 2018 | 5 | 1 | 20 | Shelter | Nasal discharge |
| 9 | Chengdu | 2018 | 6 | 2 | 33.3 | Shelter | Nasal discharge |
| 10 | Chengdu | 2018 | 4 | 2 | 20 | Shelter | Nasal discharge |
| 11 | Chongqing | 2018 | 3 | 2 | 66.7 | Shelter | Sneeze; nasal discharge |
| 12 | Chengdu | 2019 | 3 | 1 | 33.3 | Shelter | Nasal discharge |
| 13 | Chengdu | 2019 | 5 | 0 | 0 | Shelter | Sneeze; conjunctivitis |
| 14 | Chongqing | 2019 | 4 | 0 | 0 | Shelter | Sneeze; conjunctivitis |
| 15 | Chongqing | 2020 | 20 | 2 | 10 | Pet Hospital | Conjunctivitis; nasal discharge |
| 16 | Chengdu | 2020 | 58 | 6 | 10.3 | Pet Hospital | Conjunctivitis; nasal discharge |
| Total | - | - | 162 | 38 | 23.46 | - | - |
| Neutralizing Antibody (NA) | FCV Isolates | ||
|---|---|---|---|
| SMU-F4-2020 | SMU-B22-2020 | SMU-B5-2020 | |
| SMU-F4-2020-NA | 1:775 ± 65 a | 1:21.5 ± 2.5 | 1:19 ± 0 |
| SMU-B22-2020-NA | 1:106.6 ± 0.5 | 1:655 ± 55 | 1:63.5 ± 0.5 |
| SMU-B5-2020-NA | 1:95 ± 5 | 1:54.5 ± 9.5 | 1:416.5 ± 7.5 |
| Vaccine-NA | 1:16 ± 0 | 1:16 ± 0 | 1:16 ± 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Fu, N.; Ding, L.; Li, Y.; Huang, J.; Sha, X.; Zhou, Q.; Song, X.; Zhang, B. Molecular Characterization and Cross-Reactivity of Feline Calicivirus Circulating in Southwestern China. Viruses 2021, 13, 1812. https://doi.org/10.3390/v13091812
Zhou L, Fu N, Ding L, Li Y, Huang J, Sha X, Zhou Q, Song X, Zhang B. Molecular Characterization and Cross-Reactivity of Feline Calicivirus Circulating in Southwestern China. Viruses. 2021; 13(9):1812. https://doi.org/10.3390/v13091812
Chicago/Turabian StyleZhou, Long, Nengsheng Fu, Lu Ding, Yan Li, Jian Huang, Xue Sha, Qun Zhou, Xin Song, and Bin Zhang. 2021. "Molecular Characterization and Cross-Reactivity of Feline Calicivirus Circulating in Southwestern China" Viruses 13, no. 9: 1812. https://doi.org/10.3390/v13091812
APA StyleZhou, L., Fu, N., Ding, L., Li, Y., Huang, J., Sha, X., Zhou, Q., Song, X., & Zhang, B. (2021). Molecular Characterization and Cross-Reactivity of Feline Calicivirus Circulating in Southwestern China. Viruses, 13(9), 1812. https://doi.org/10.3390/v13091812