
High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Retrieval
2.2. Data Processing
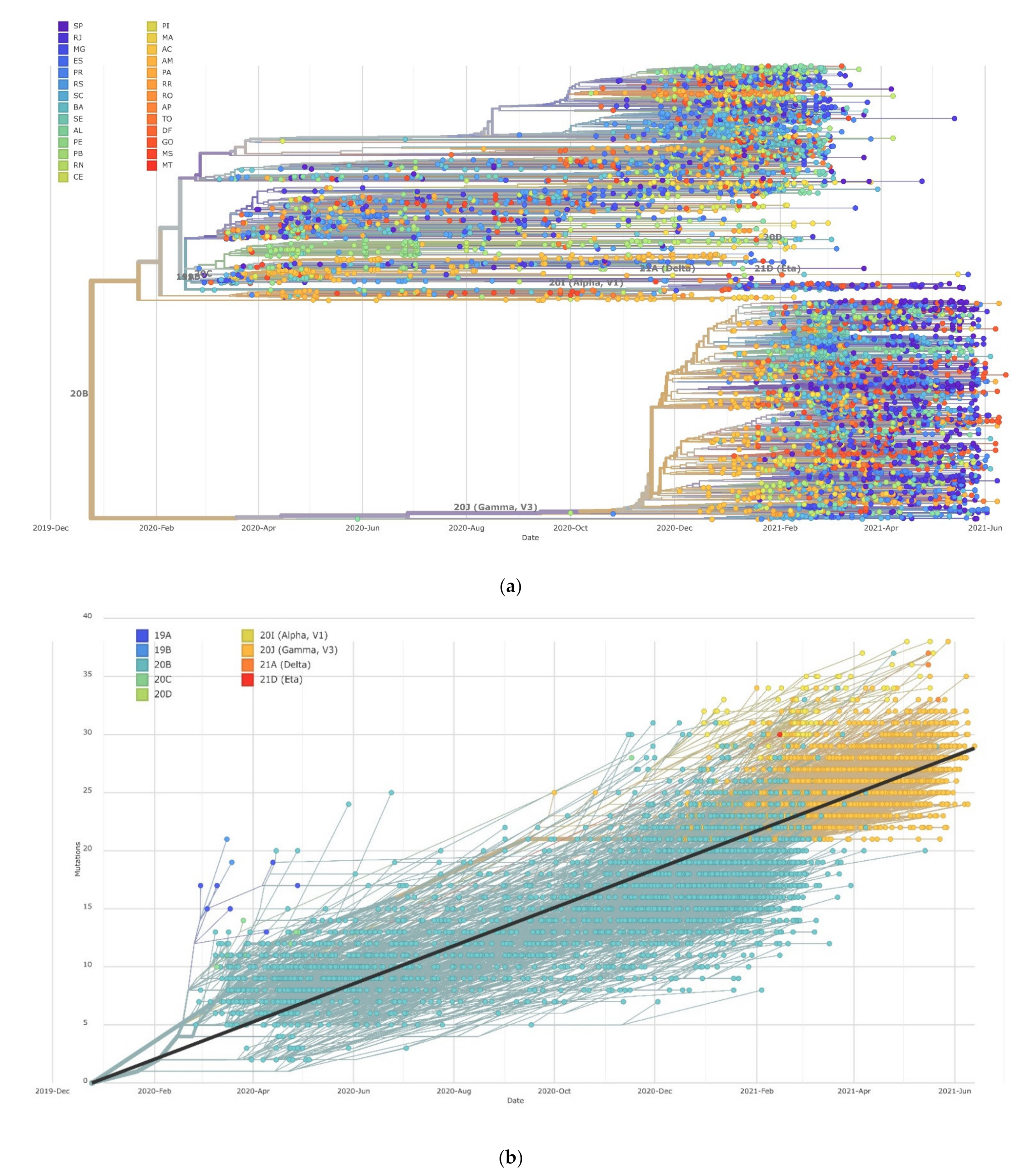
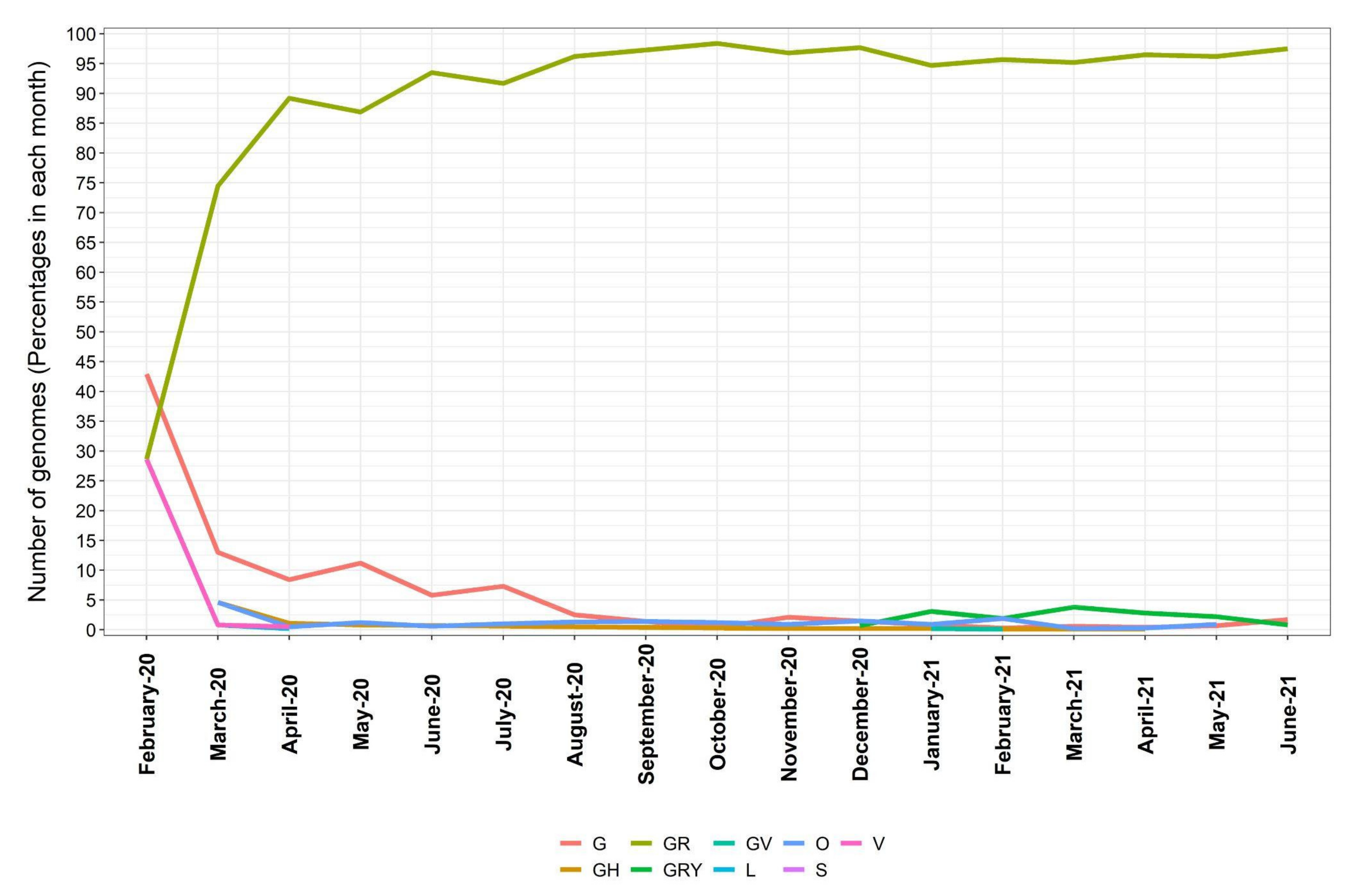
2.3. Dynamics of SARS-CoV Clades
2.4. Mathematical Model to Estimate the Rate of Genome Mutants and Global Diversity Rate
3. Results
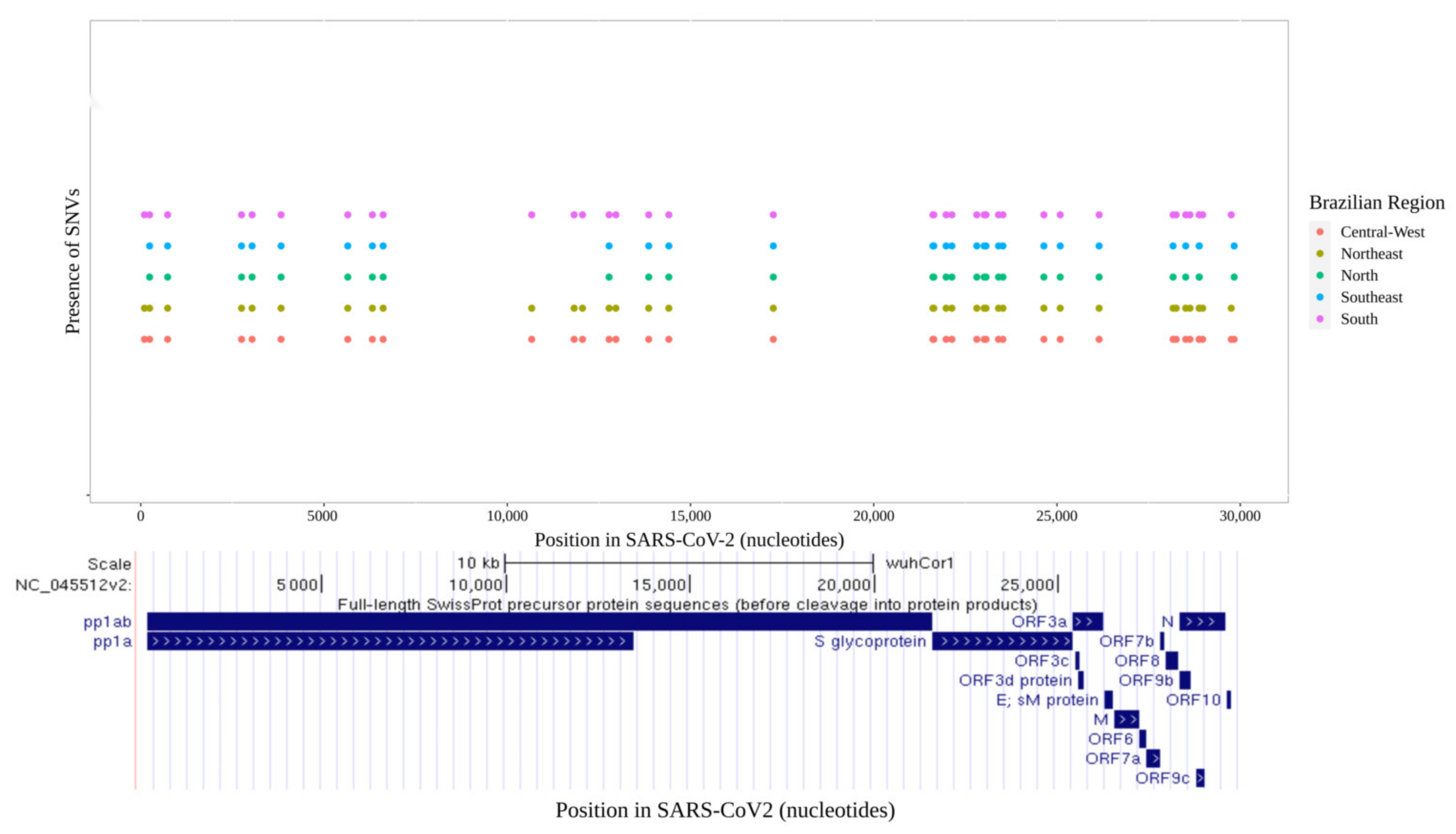
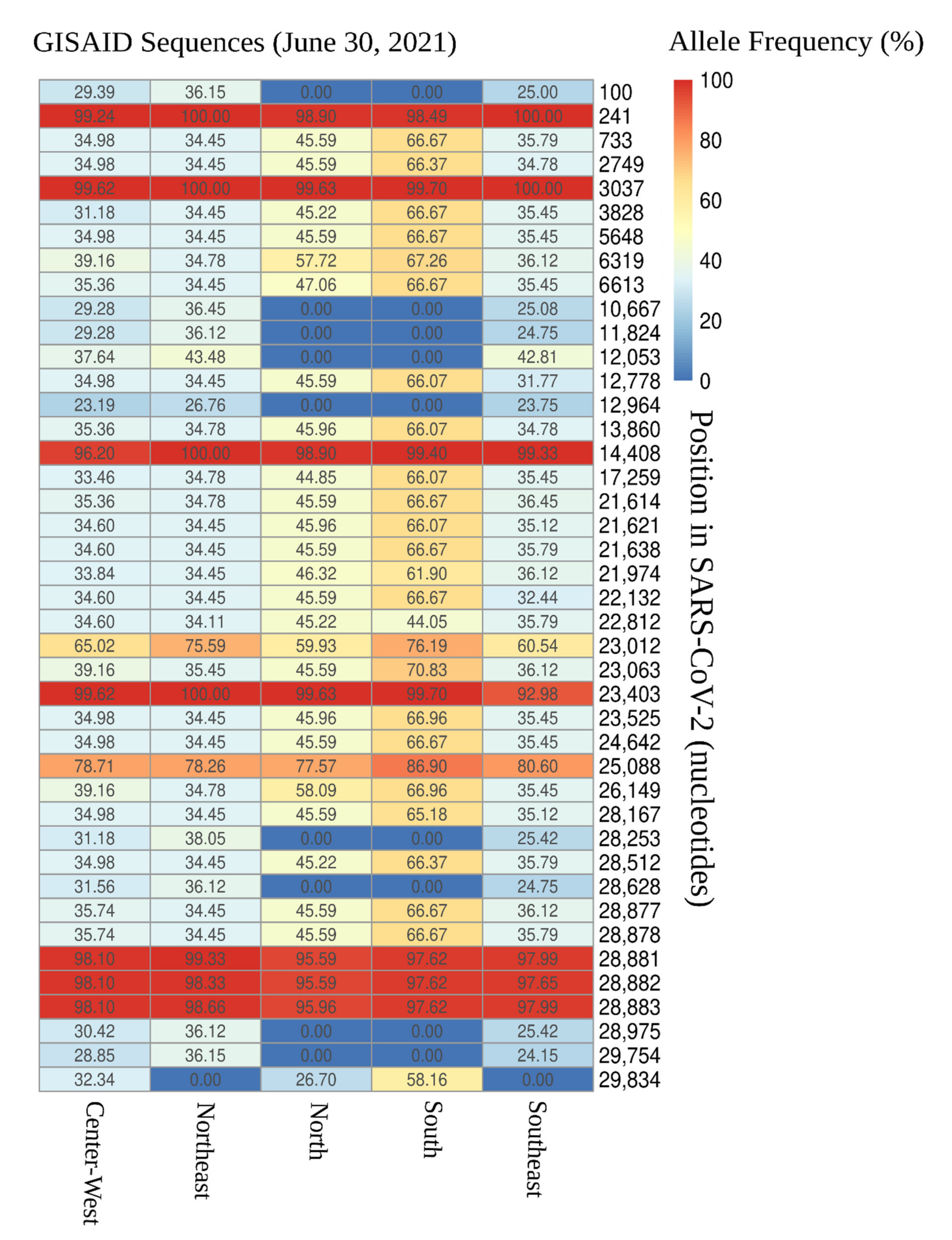
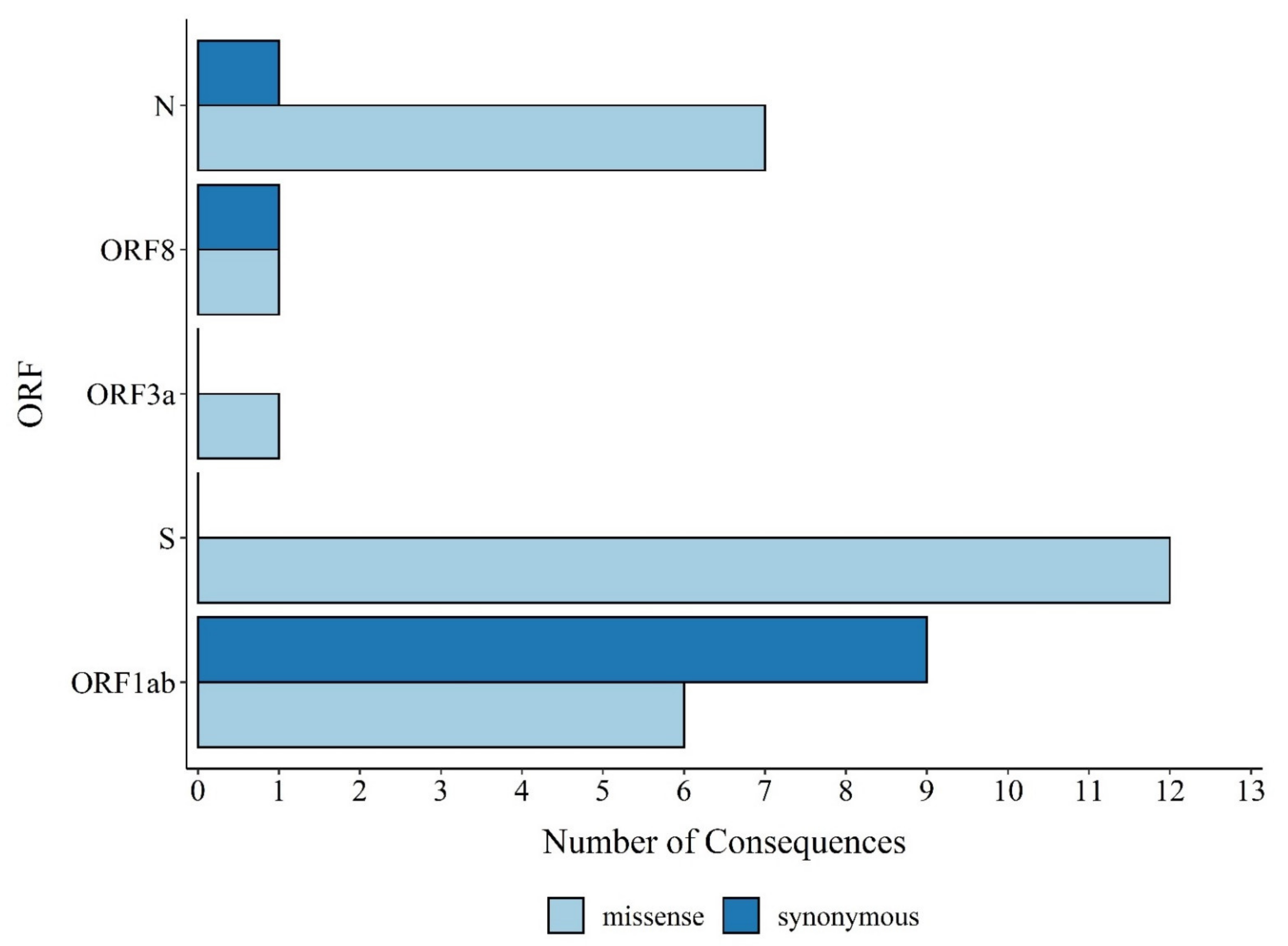
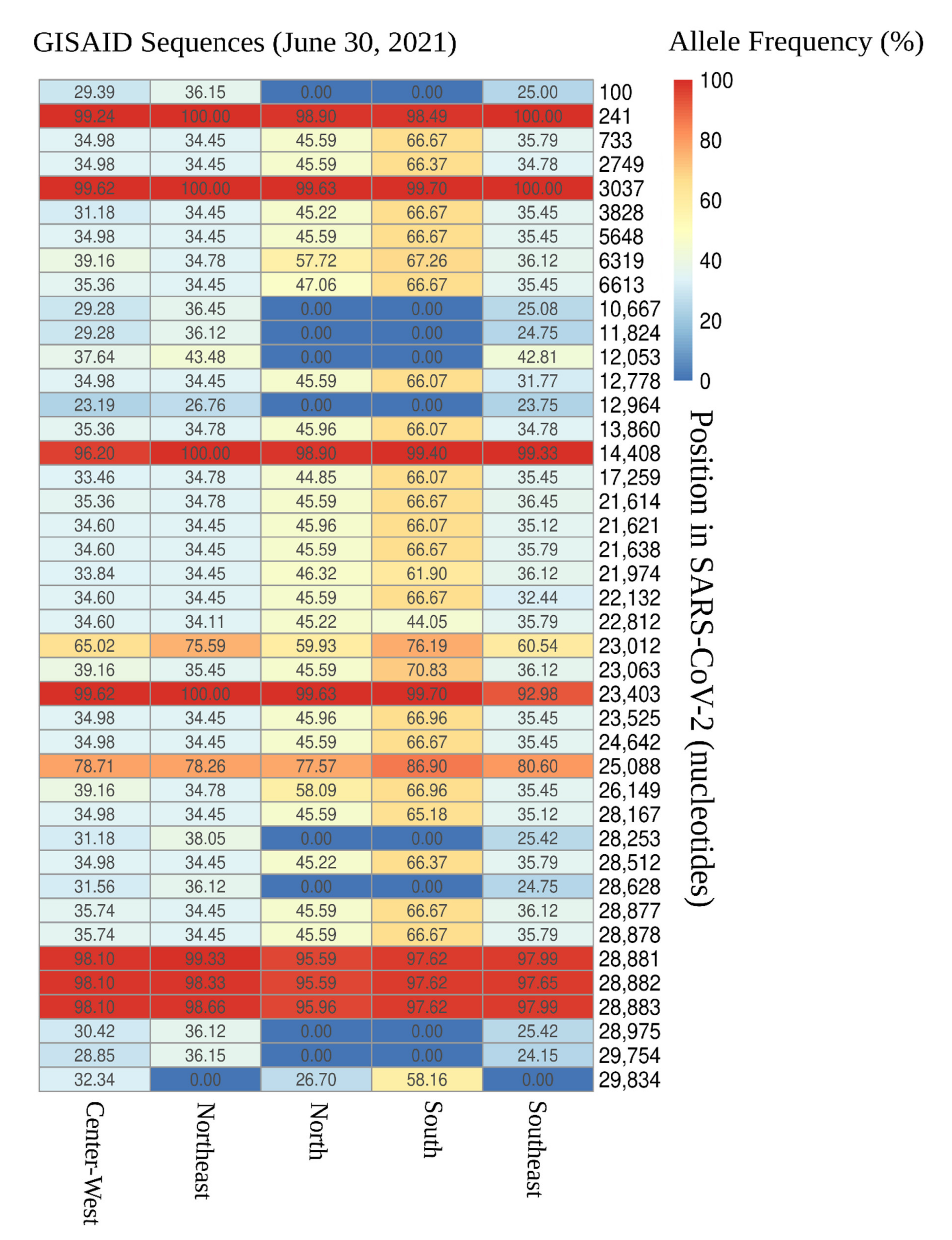
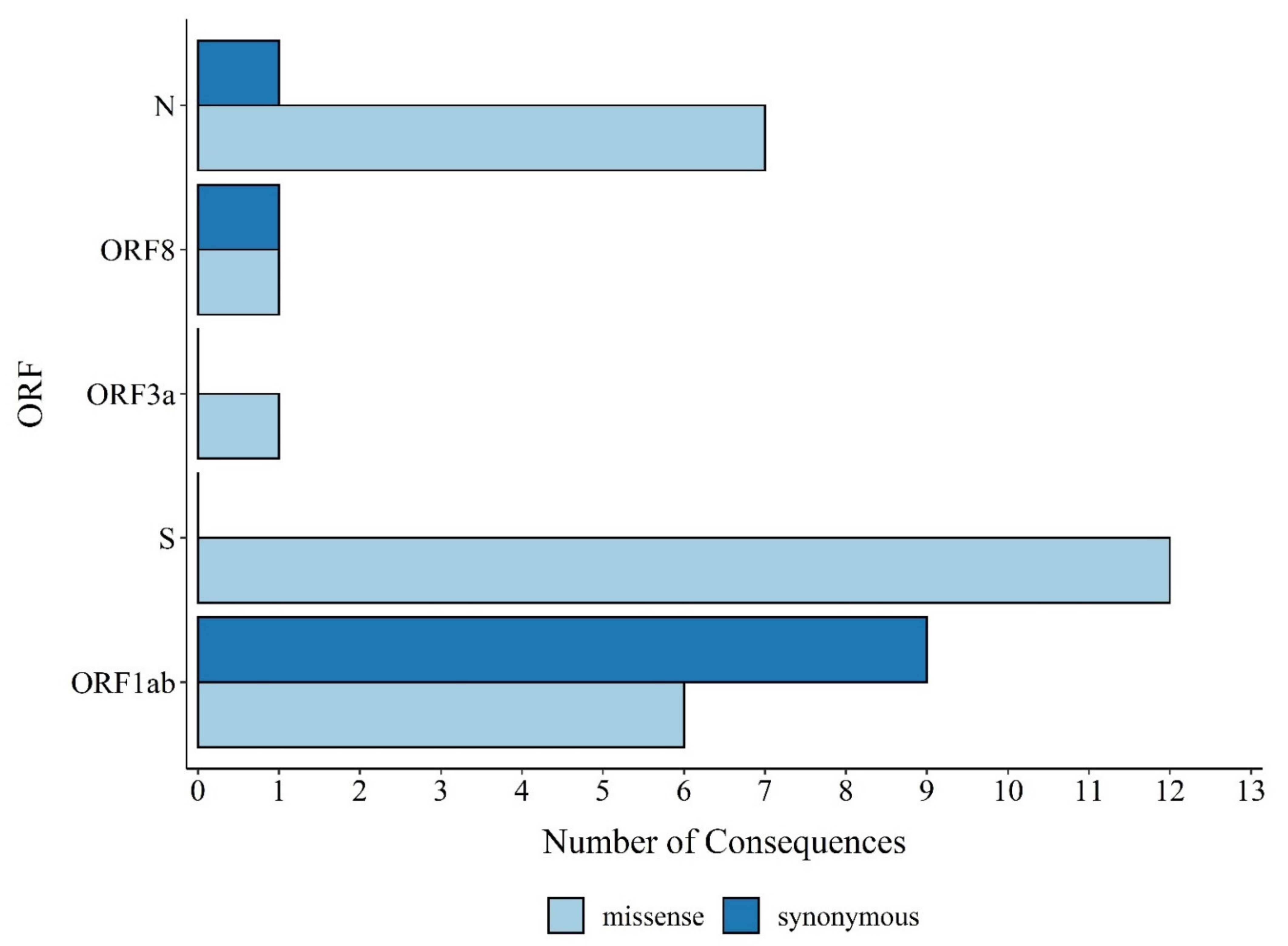
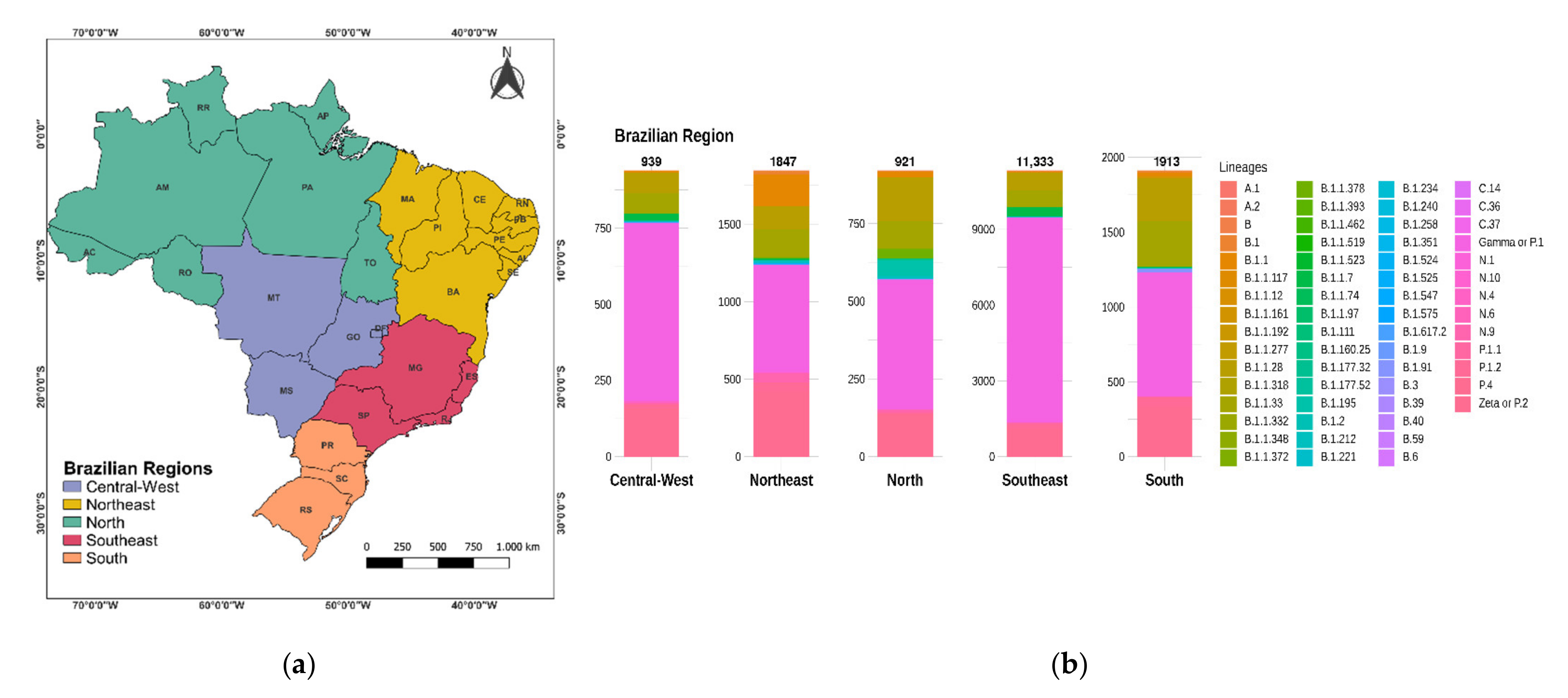
3.1. Distribution of SARS-CoV-2 Mutations in Brazil
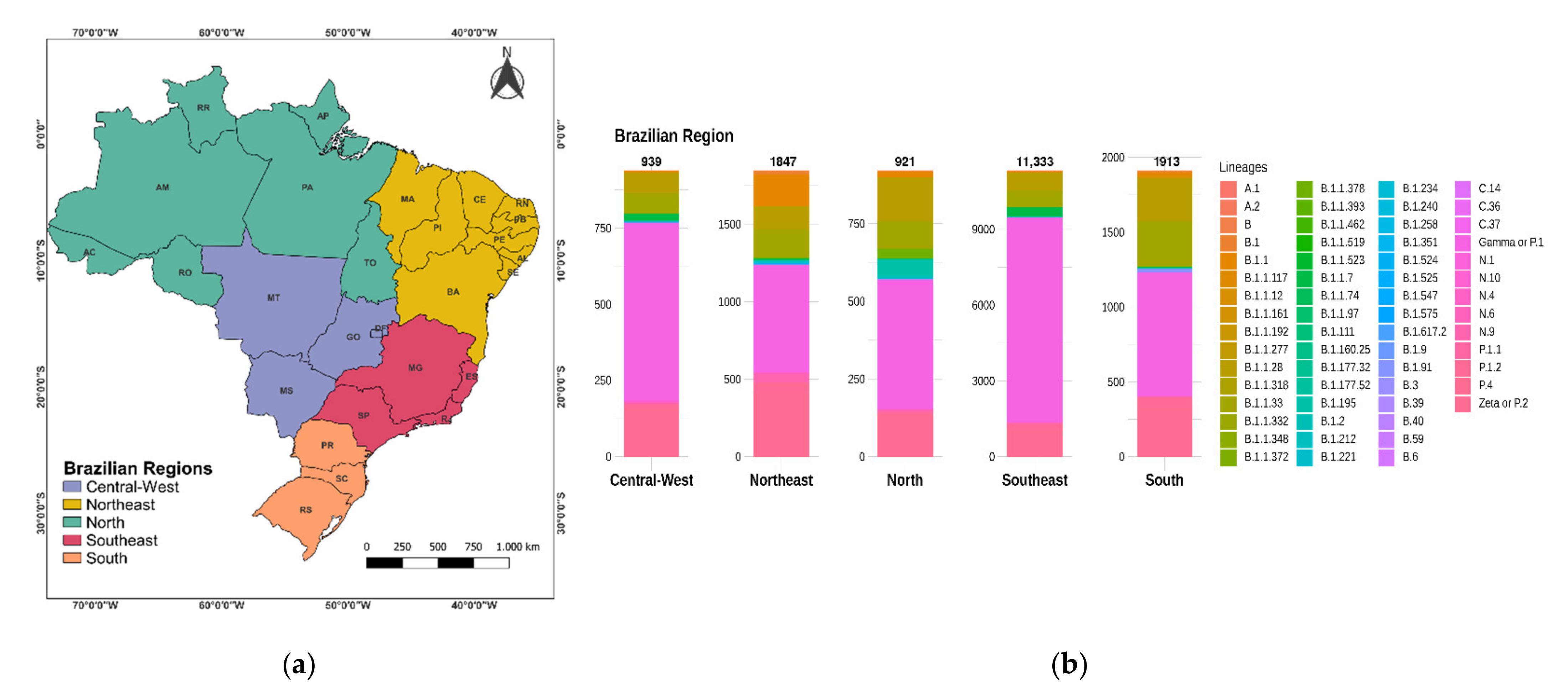
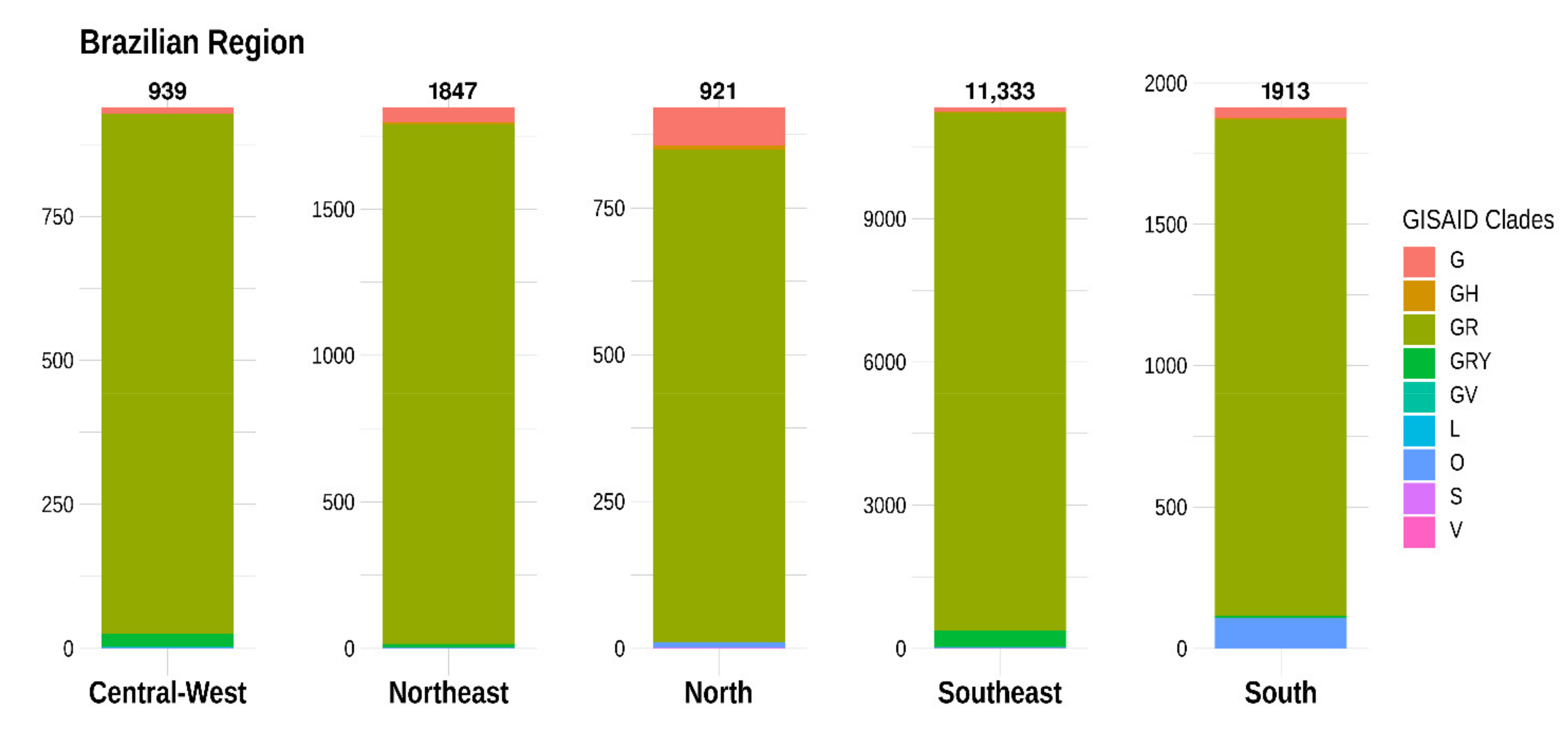
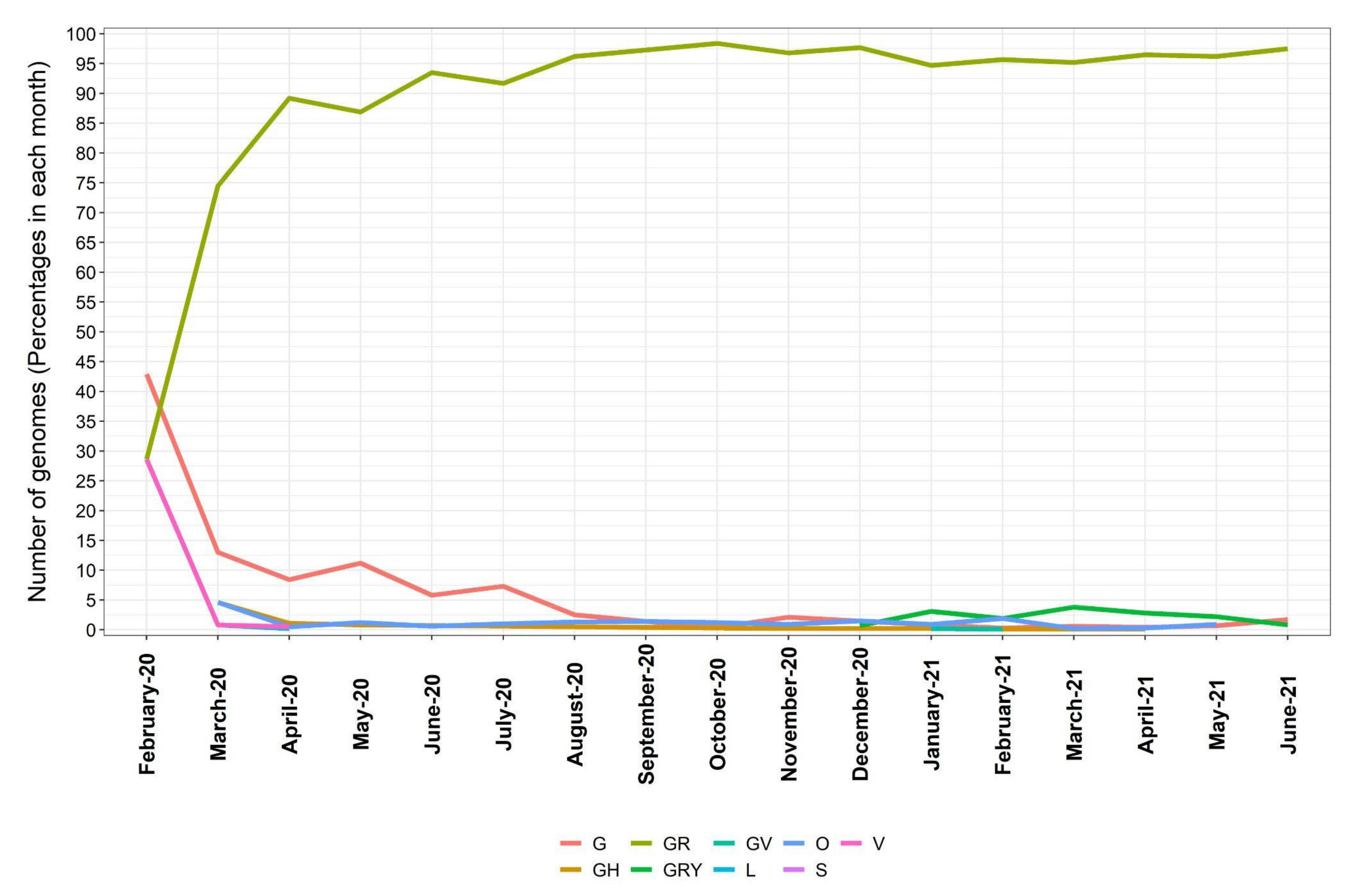
3.2. Distribution of SARS-CoV-2 Clades and Lineages/Variants in Brazil
3.3. Numbers of Genomes to Identify a New Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 8 July 2021).
- Deng, W.; Bao, L.; Liu, J.; Xiao, C.; Liu, J.; Xue, J.; Lv, Q.; Qi, F.; Gao, H.; Yu, P.; et al. Primary exposure to SARS-CoV-2 protects against reinfection in rhesus macaques. Science 2020, 369, 818–823. [Google Scholar] [CrossRef]
- Holmes, E.C.; Rambaut, A. Viral evolution and the emergence of SARS coronavirus. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004, 359, 1059–1065. [Google Scholar] [CrossRef] [Green Version]
- Plowright, R.K.; Parrish, C.R.; McCallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhshandeh, B.; Sorboni, S.G.; Javanmard, A.-R.; Mottaghi, S.S.; Mehrabi, M.-R.; Sorouri, F.; Abbasi, A.; Jahanafrooz, Z. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect. Genet. Evol. 2021, 90, 104773. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Lima, L.B.; Montenegro, R.C. The human pandemic coronaviruses on the show: The spike glycoprotein as the main actor in the coronaviruses play. Int. J. Biol. Macromol. 2021, 179, 1–19. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–882. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nat. Cell Biol. 2021, 595, 707–712. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nat. Cell Biol. 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wu, L.J.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.N.; Lou, Z.Y.; Yan, L.M.; Zhang, R.G.; Rao, Z.H. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guruprasad, L. Human coronavirus spike protein-host receptor recognition. Prog. Biophys. Mol. Biol. 2021, 161, 39–53. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. bioRxiv 2020. Update in: Cell 2020,183, 739–751.e8. Available online: https://pubmed.ncbi.nlm.nih.gov/32637944/ (accessed on 1 August 2021). [CrossRef]
- Shu, Y.; McCauley, J. GISAID:The cr Global initiative on sharing all influenza data-from vision to reality. Eur. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Hamed, S.M.; Elkhatib, W.F.; Khairalla, A.S.; Noreddin, A.M. Global dynamics of SARS-CoV-2 clades and their relation to COVID-19 epidemiology. Sci. Rep. 2021, 11, 8435. [Google Scholar] [CrossRef]
- Cave, E. COVID-19 Super-spreaders: Definitional Quandaries and Implications. Asian Bioeth. Rev. 2020, 12, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Rouchka, E.C.; Chariker, J.H.; Chung, D. Variant analysis of 1,040 SARS-CoV-2 genomes. PLoS ONE 2020, 15, e0241535. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Resende, P.C.; Bezerra, J.F.; de Vasconcelos, R.H.T.; Arantes, I.; Appolinario, L.; Mendonça, A.C.; Paixao, A.C.; Rodrigues, A.C.D.; Silva, T.; Rocha, A.S.; et al. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020-SARS-CoV-2 coronavirus. Virol. org. 2021. Available online: https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584 (accessed on 7 July 2021).
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Gonçalves, L.; et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein-SARS-CoV-2 coronavirus/nCoV-2019. Virol. Org. 2021. Available online: https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spike-protein/585 (accessed on 7 July 2021).
- Motayo, B.O.; Oluwasemowo, O.O.; Olusola, B.; Akinduti, P.A.; Arege, O.T.; Obafemi, Y.D.; Faneye, A.O.; Isibor, P.O.; Aworunse, O.S.; Oranusi, S.U. Evolution and genetic diversity of SARS-CoV-2 in Africa using whole genome sequences. Int. J. Infect. Dis. 2021, 103, 282–287. [Google Scholar] [CrossRef]
- Nonaka, C.K.V.; Franco, M.M.; Gräf, T.; Barcia, C.A.d.; Mendonça, R.N.d.; de Sousa, K.A.F.; Neiva, L.M.C.; Fosenca, V.; Mendes, A.V.A.; de Aguiar, R.S.; et al. Genomic Evidence of SARS-CoV-2 Reinfection Involving E484K Spike Mutation, Brazil. Emerg. Infect. Dis. 2021, 27, 1522–1524. [Google Scholar] [CrossRef]
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New insights into genetic susceptibility of COVID-19: An ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020, 18, 216. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | n of Genomes | n Max of Lineages | G/L * | n of Countries | Diversity Region Index |
|---|---|---|---|---|---|
| South America | 26,257 | 189 | 25.28 | 15 | 3.6906 |
| Oceania | 15,484 | 249 | 24.85 | 6 | 0.9893 |
| Europe | 1,000,285 | 956 | 293.21 | 49 | 8.4996 |
| Asia | 112,901 | 512 | 49.01 | 38 | 5.9559 |
| North America | 477,842 | 732 | 188.30 | 17 | 1.6557 |
| Africa | 11,873 | 239 | 13.99 | 38 | 6.4225 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, U.J.B.; dos Santos, R.N.; Campos, F.S.; Lourenço, K.L.; da Fonseca, F.G.; Spilki, F.R.; Corona-ômica.BR/MCTI Network. High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses 2021, 13, 1806. https://doi.org/10.3390/v13091806
de Souza UJB, dos Santos RN, Campos FS, Lourenço KL, da Fonseca FG, Spilki FR, Corona-ômica.BR/MCTI Network. High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses. 2021; 13(9):1806. https://doi.org/10.3390/v13091806
Chicago/Turabian Stylede Souza, Ueric José Borges, Raíssa Nunes dos Santos, Fabrício Souza Campos, Karine Lima Lourenço, Flavio Guimarães da Fonseca, Fernando Rosado Spilki, and Corona-ômica.BR/MCTI Network. 2021. "High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021" Viruses 13, no. 9: 1806. https://doi.org/10.3390/v13091806
APA Stylede Souza, U. J. B., dos Santos, R. N., Campos, F. S., Lourenço, K. L., da Fonseca, F. G., Spilki, F. R., & Corona-ômica.BR/MCTI Network. (2021). High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses, 13(9), 1806. https://doi.org/10.3390/v13091806