
Large Evolutionary Rate Heterogeneity among and within HIV-1 Subtypes and CRFs


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Retrieval
2.2. Data Filtering and Quality Control
2.3. Dataset Sub-Sampling
2.4. Phylogenetic Tree Reconstruction
2.5. Phylogenetic Tree Dating
2.6. Tip-Date Randomization Test
2.7. Rate Autocorrelation Test
2.8. Leave-One-Out (LOO) Cross-Validation
2.9. Exploration of Population Structure in Time-Scaled Phylogenies
3. Results
3.1. Substantial Substitution Rate Heterogeneity between HIV-1 Subtypes and CRFs
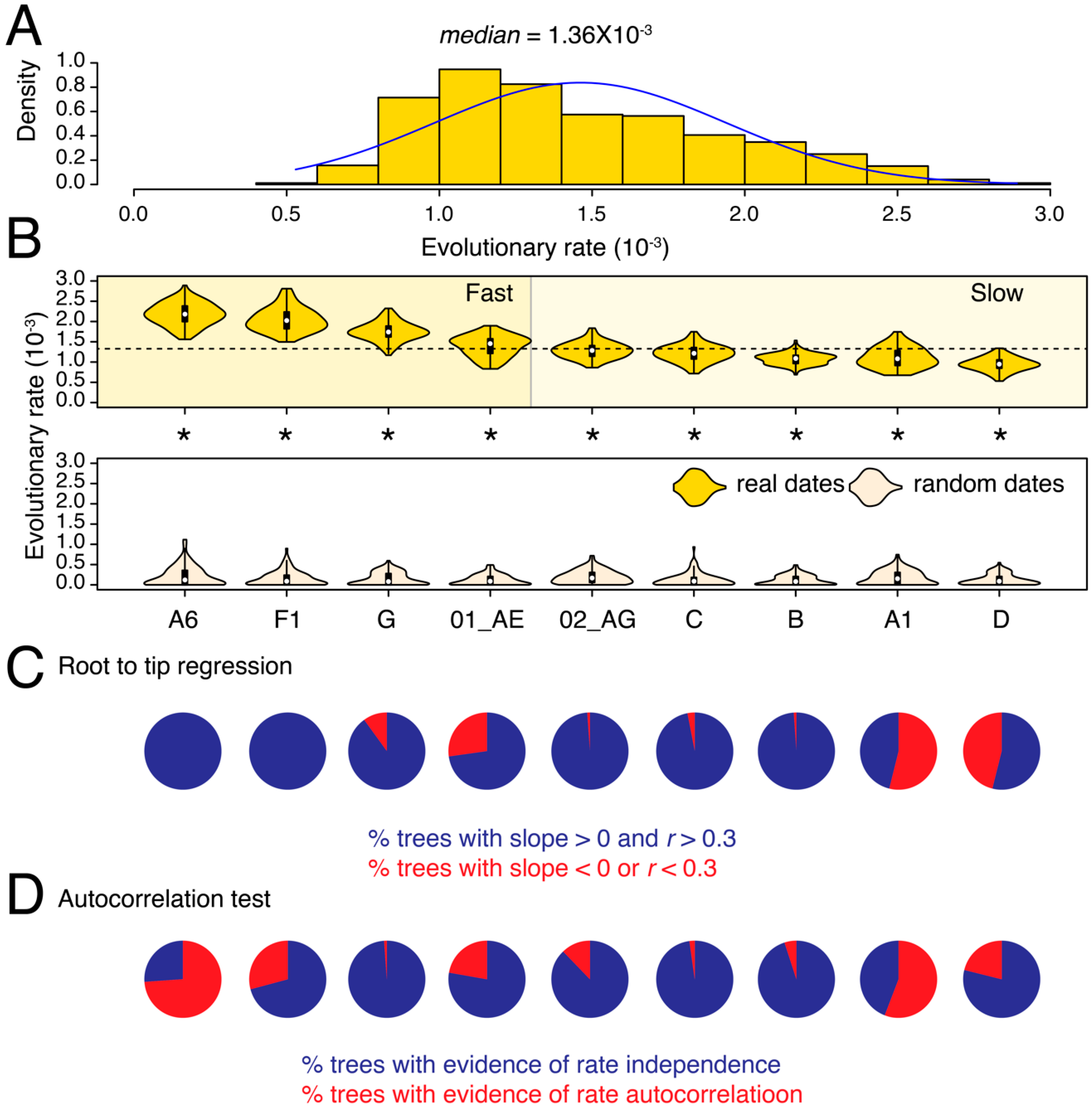
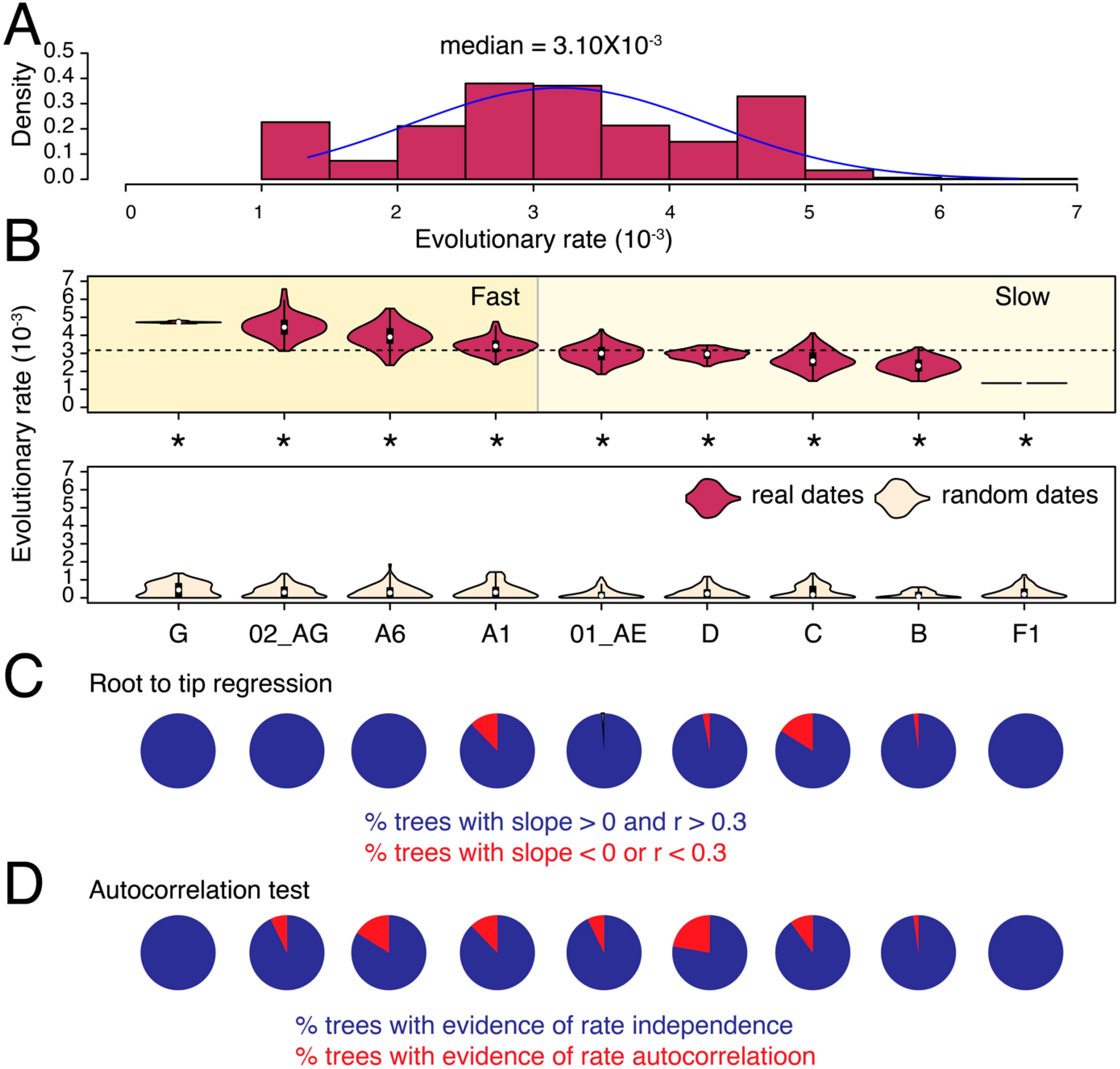

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CorrTest | Root-to-Tip Regression | |||
|---|---|---|---|---|
| Subtype/CRF | Autocorrelated | Independent | Pass | Fail |
| env | ||||
| 01_AE | 7 | 93 | 99 | 1 |
| 02_AG | 7 | 93 | 100 | 0 |
| A1 | 12 | 88 | 88 | 12 |
| A6 | 16 | 84 | 100 | 0 |
| B | 2 | 98 | 98 | 2 |
| C | 10 | 90 | 84 | 16 |
| D | 22 | 78 | 97 | 3 |
| F1 | 0 | 100 | 100 | 0 |
| G | 0 | 100 | 100 | 0 |
| pol | ||||
| 01_AE | 22 | 78 | 73 | 27 |
| 02_AG | 12 | 88 | 99 | 1 |
| A1 | 56 | 44 | 46 | 54 |
| A6 | 74 | 26 | 100 | 0 |
| B | 5 | 95 | 99 | 1 |
| C | 2 | 98 | 97 | 3 |
| D | 21 | 79 | 54 | 46 |
| F1 | 29 | 71 | 100 | 0 |
| G | 1 | 99 | 90 | 10 |
| Subtype/CRF | Gene | Trees | Mean | Median | Std. Error | Lower-Bound | Upper-Bound |
|---|---|---|---|---|---|---|---|
| G | env | 100 | 4.72 | 4.72 | 0.0045 | 4.71 | 4.73 |
| 02_AG | env | 100 | 4.48 | 4.45 | 0.0623 | 4.36 | 4.60 |
| A6 | env | 100 | 3.94 | 3.91 | 0.0647 | 3.81 | 4.06 |
| A1 | env | 88 | 3.41 | 3.41 | 0.0461 | 3.32 | 3.50 |
| 01_AE | env | 99 | 2.99 | 3.02 | 0.047 | 2.90 | 3.09 |
| D | env | 97 | 2.93 | 2.98 | 0.0261 | 2.88 | 2.98 |
| C | env | 84 | 2.74 | 2.64 | 0.0555 | 2.63 | 2.85 |
| B | env | 98 | 2.33 | 2.32 | 0.0418 | 2.25 | 2.41 |
| F1 | env | 100 | 1.34 | 1.34 | 0.00003 | 1.34 | 1.34 |
| A6 | pol | 100 | 2.18 | 2.18 | 0.0275 | 2.12 | 2.23 |
| F1 | pol | 100 | 2.05 | 2.02 | 0.0294 | 1.99 | 2.11 |
| G | pol | 90 | 1.78 | 1.76 | 0.0225 | 1.73 | 1.82 |
| 01_AE | pol | 73 | 1.48 | 1.51 | 0.0238 | 1.43 | 1.52 |
| 02_AG | pol | 99 | 1.29 | 1.29 | 0.0200 | 1.25 | 1.33 |
| C | pol | 97 | 1.22 | 1.23 | 0.0206 | 1.18 | 1.26 |
| B | pol | 99 | 1.08 | 1.09 | 0.014 | 1.05 | 1.11 |
| A1 | pol | 46 | 1.25 | 1.26 | 0.0338 | 1.18 | 1.31 |
| D | pol | 54 | 0.98 | 0.96 | 0.0200 | 0.94 | 1.02 |
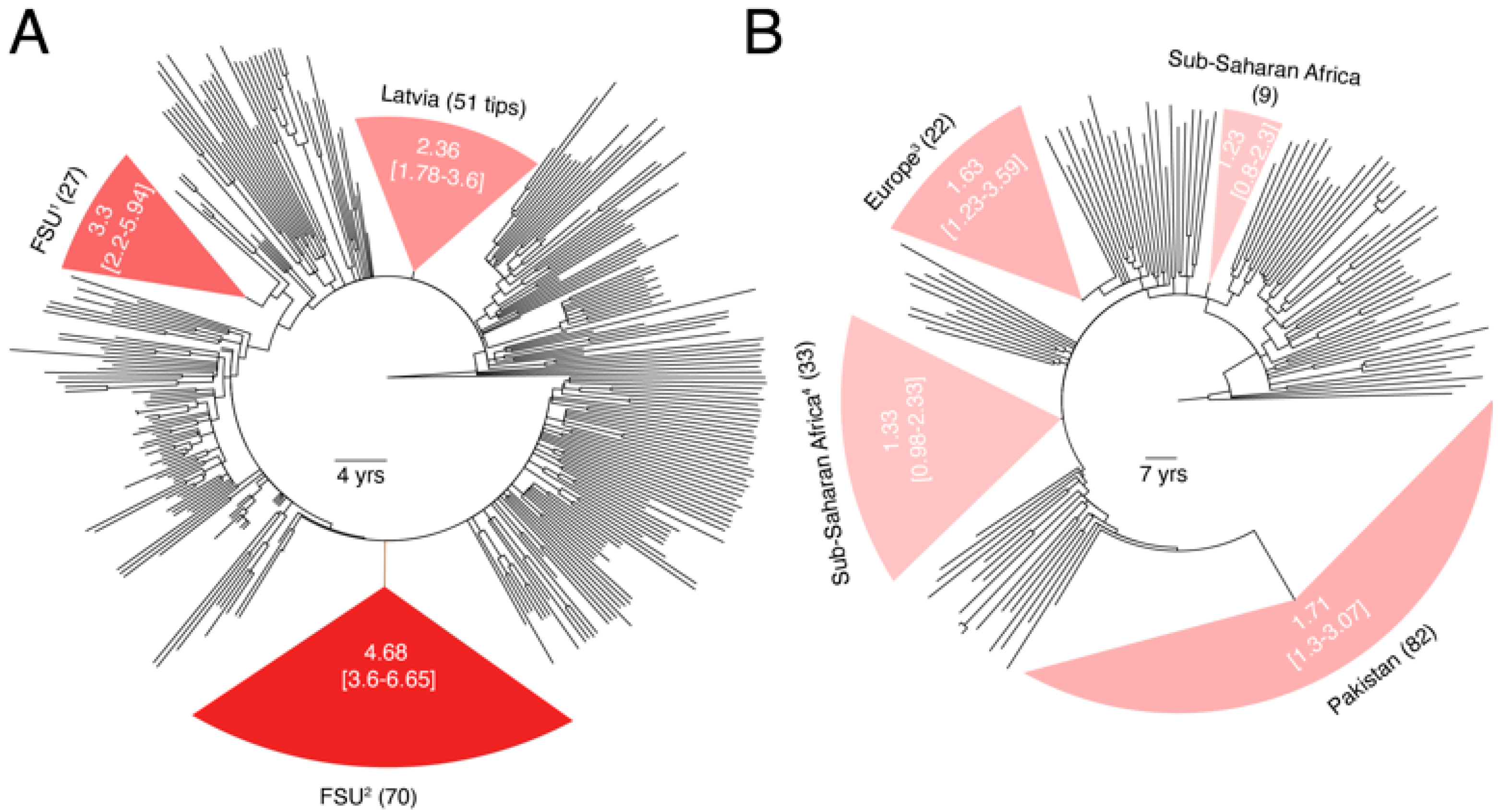
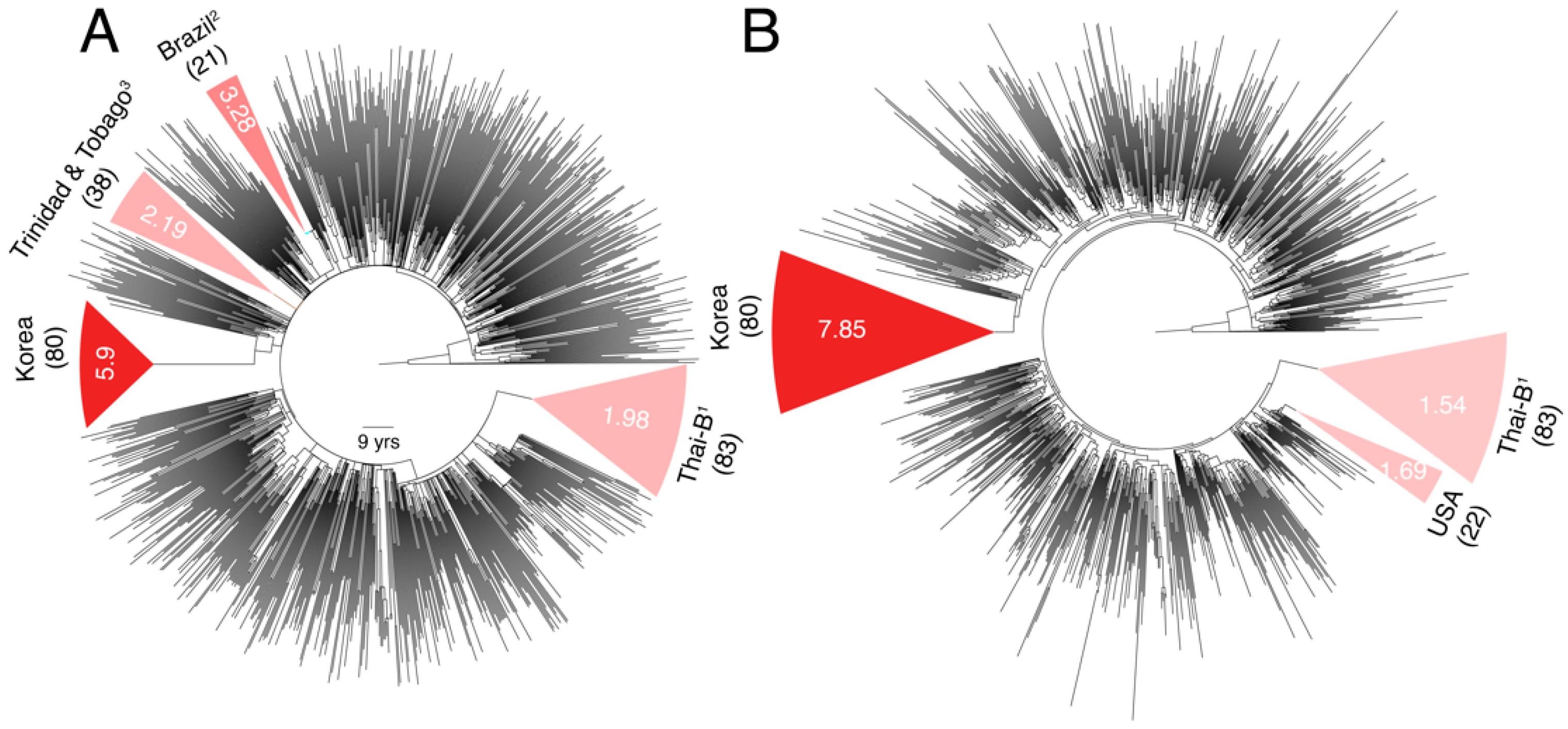
3.2. Substantial Substitution Rate Heterogeneity between Closely-Related HIV-1 Sub-Subtypes
3.3. Substantial Substitution Rate Heterogeneity within Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—Fact Sheet. 2021. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 19 August 2021).
- Frank, T.D.; Carter, A.; Jahagirdar, D.; Biehl, M.H.; Douwes-Schultz, D.; Larson, S.L.; Arora, M.; Dwyer-Lindgren, L.; Steuben, K.M.; Abbastabar, H.; et al. Global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2017, and forecasts to 2030, for 195 countries and territories: A systematic analysis for the Global Burden of Diseases, Injuries, and Risk Factors Study 2017. Lancet HIV 2019, 6, e831–e859. [Google Scholar] [CrossRef] [Green Version]
- Dwyer-Lindgren, L.; Cork, M.A.; Sligar, A.; Steuben, K.M.; Wilson, K.F.; Provost, N.R.; Mayala, B.K.; VanderHeide, J.D.; Collison, M.L.; Hall, J.B.; et al. Mapping HIV prevalence in sub-Saharan Africa between 2000 and 2017. Nature 2019, 570, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Burton, D.R. Advancing an HIV vaccine; advancing vaccinology. Nat. Rev. Immunol. 2019, 19, 77–78. [Google Scholar] [CrossRef]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [CrossRef] [Green Version]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Leitner, T. The puzzle of HIV neutral and selective evolution. Mol. Biol. Evol. 2018, 35, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Kirtley, S.; Gouws-Williams, E.; Ghys, P.D.; Abimiku, A.G.; Agwale, S.; Archibald, C.; et al. Global and regional epidemiology of HIV-1 recombinants in 1990–2015: A systematic review and global survey. Lancet HIV 2020, 7, e772–e781. [Google Scholar] [CrossRef]
- Alizon, S.; Fraser, C. Within-host and between-host evolutionary rates across the HIV-1 genome. Retrovirology 2013, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Raghwani, J.; Redd, A.D.; Longosz, A.F.; Wu, C.H.; Serwadda, D.; Martens, C.; Kagaayi, J.; Sewankambo, N.; Porcella, S.F.; Grabowski, M.K.; et al. Evolution of HIV-1 within untreated individuals and at the population scale in Uganda. PLoS Pathog. 2018, 14, e1007167. [Google Scholar] [CrossRef] [Green Version]
- Leitner, T. Phylogenetics in HIV transmission: Taking within-host diversity into account. Curr. Opin. HIV AIDS 2019, 14, 181–187. [Google Scholar] [CrossRef]
- Berry, I.M.; Athreya, G.; Kothari, M.; Daniels, M.; Bruno, W.J.; Korber, B.; Kuiken, C.; Ribeiro, R.M.; Leitner, T. The evolutionary rate dynamically tracks changes in HIV-1 epidemics: Application of a simple method for optimizing the evolutionary rate in phylogenetic trees with longitudinal data. Epidemics 2009, 1, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Berry, I.M.; Ribeiro, R.; Kothari, M.; Athreya, G.; Daniels, M.; Lee, H.Y.; Bruno, W.; Leitner, T. Unequal evolutionary rates in the human immunodeficiency virus type 1 (HIV-1) pandemic: The evolutionary rate of HIV-1 slows down when the epidemic rate increases. J. Virol. 2007, 81, 10625–10635. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.; Poon, A.F.Y. Phylogenetic measures of indel rate variation among the HIV-1 group M subtypes. Virus Evol. 2019, 5, vez022. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, J.O.; Fourment, M.; Kosakovsky Pond, S.L. Inconsistencies in estimating the age of HIV-1 subtypes due to heterotachy. Mol. Biol. Evol. 2012, 29, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Foley, B.T.; Korber, B.T.M.; Leitner, T.K.; Apetrei, C.; Beatrice, H.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S. HIV Sequence Compendium 2018; Theoretical Biology and Biophysics, Los Alamos National Laboratory: Los Alamos, NM, USA, 2018; LA-UR-18-25673.
- Leitner, T.; Escanilla, D.; Franzén, C.; Uhlén, M.; Albert, J. Accurate reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proc. Natl. Acad. Sci. USA 1996, 93, 10864. [Google Scholar] [CrossRef] [Green Version]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef]
- Junqueira, D.M.; de Matos Almeida, S.E. HIV-1 subtype B: Traces of a pandemic. Virology 2016, 495, 173–184. [Google Scholar] [CrossRef]
- Miura, S.; Tamura, K.; Tao, Q.; Huuki, L.A.; Kosakovsky Pond, S.L.; Priest, J.; Deng, J.; Kumar, S. A new method for inferring timetrees from temporally sampled molecular sequences. PLoS Comput. Biol. 2020, 16, e1007046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, T.H.; Jung, M.; Lycett, S.; Gascuel, O. Fast dating using least-squares criteria and algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Tao, Q.; Tamura, K.U.; Battistuzzi, F.; Kumar, S. A machine learning method for detecting autocorrelation of evolutionary rates in large phylogenies. Mol. Biol. Evol. 2019, 36, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; Calcagnì, A. Measuring distribution similarities between samples: A distribution-free overlapping index. Front. Psychol. 2019, 10, 1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volz, E.M.; Carsten, W.; Grad, Y.H.; Frost, S.D.W.; Dennis, A.M.; Didelot, X. Identification of hidden population structure in time-scaled phylogenies. Syst. Biol. 2020, 69, 884–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, A.B.; Vandamme, A.-M.; Lemey, P. Quantifying differences in the tempo of human immunodeficiency virus type 1 subtype evolution. J. Virol. 2009, 83, 12917–12924. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.J.; Yun, M.R.; Koo, M.J.; Shin, B.G.; Lee, J.S.; Kim, S.S. Estimating the origin and evolution characteristics for Korean HIV type 1 subtype B using Bayesian phylogenetic analysis. AIDS Res. Hum. Retrovir. 2012, 28, 880–884. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Choi, B.Y.; Kim, S.M.; Kim, S., II; Kim, J.; Choi, J.Y.; Kim, S.W.; Song, J.Y.; Kim, Y.J.; Park, D.W.; et al. Epidemiological characteristics of HIV infected Korean: Korea HIV/AIDS cohort study. Epidemiol. Health 2019, 41, e2019037. [Google Scholar] [CrossRef]
- Kim, G.J.; Nam, J.-G.; Shin, B.G.; Kee, M.K.; Kim, E.-J.; Lee, J.-S.; Kim, S.S. National survey of prevalent HIV strains: Limited genetic variation of Korean HIV-1 clade B within the population of Korean men who have sex with men. J. Acquir. Immune Defic. Syndr. 2008, 48, 127–132. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.Á.; González-Candelas, F. The substitution rate of HIV-1 subtypes: A genomic approach. Virus Evol. 2017, 3, vex029. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent rate phenomenon in viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchêne, S.; Holmes, E.C.; Ho, S.Y.W. Analyses of evolutionary dynamics in viruses are hindered by a time-dependent bias in rate estimates. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140732. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-dependent rates of molecular evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef]
- Albert, J.; Berglund, T.; Gisslén, M.; Gröön, P.; Sönnerborg, A.; Tegnell, A.; Alexandersson, A.; Berggren, I.; Blaxhult, A.; Brytting, M.; et al. Risk of HIV transmission from patients on antiretroviral therapy: A position statement from the public health agency of Sweden and the Swedish reference group for antiviral therapy. Scand. J. Infect. Dis. 2014, 46, 673–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasir, A.; Dimitrijevic, M.; Romero-Severson, E.; Leitner, T. Large Evolutionary Rate Heterogeneity among and within HIV-1 Subtypes and CRFs. Viruses 2021, 13, 1689. https://doi.org/10.3390/v13091689
Nasir A, Dimitrijevic M, Romero-Severson E, Leitner T. Large Evolutionary Rate Heterogeneity among and within HIV-1 Subtypes and CRFs. Viruses. 2021; 13(9):1689. https://doi.org/10.3390/v13091689
Chicago/Turabian StyleNasir, Arshan, Mira Dimitrijevic, Ethan Romero-Severson, and Thomas Leitner. 2021. "Large Evolutionary Rate Heterogeneity among and within HIV-1 Subtypes and CRFs" Viruses 13, no. 9: 1689. https://doi.org/10.3390/v13091689
APA StyleNasir, A., Dimitrijevic, M., Romero-Severson, E., & Leitner, T. (2021). Large Evolutionary Rate Heterogeneity among and within HIV-1 Subtypes and CRFs. Viruses, 13(9), 1689. https://doi.org/10.3390/v13091689