
Persistence of Multiple Paramyxoviruses in a Closed Captive Colony of Fruit Bats (Eidolon helvum)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
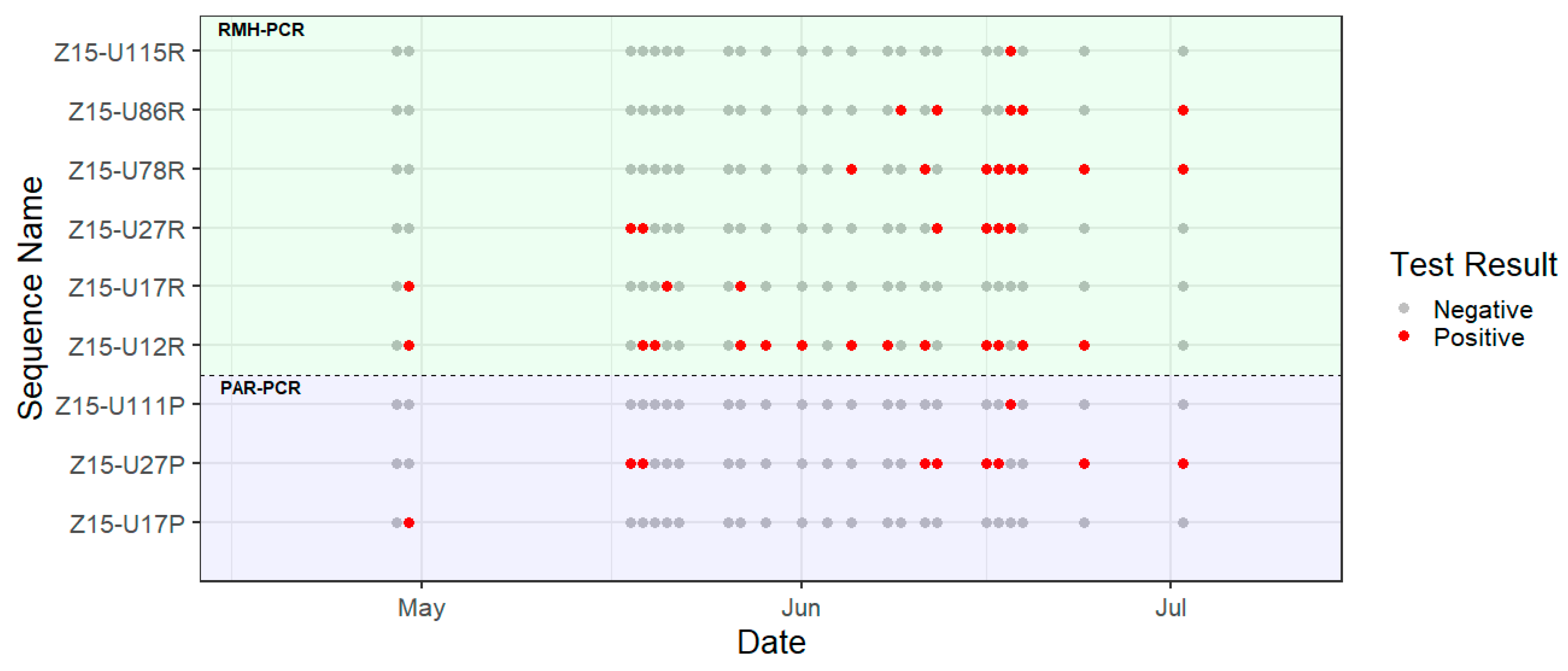
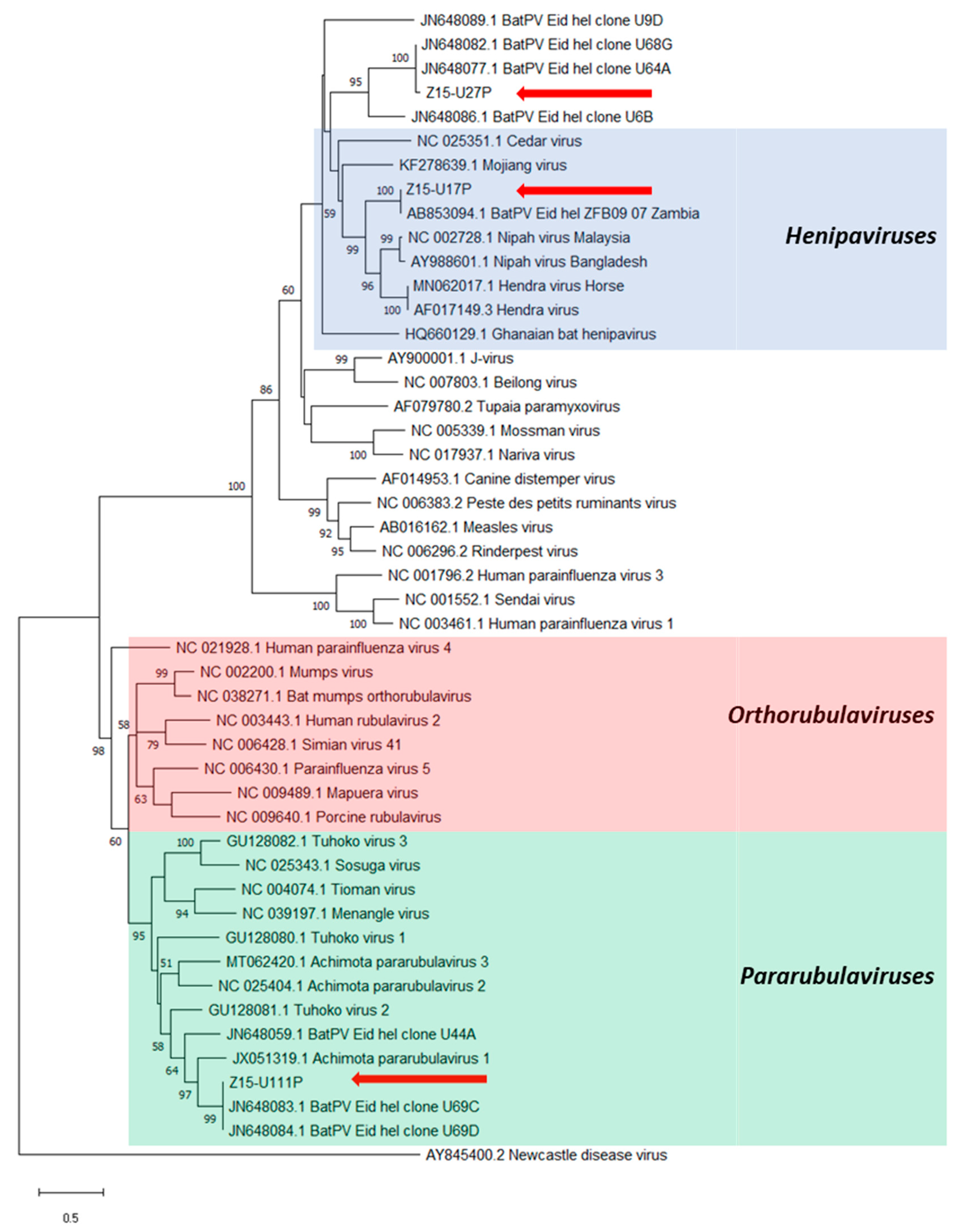
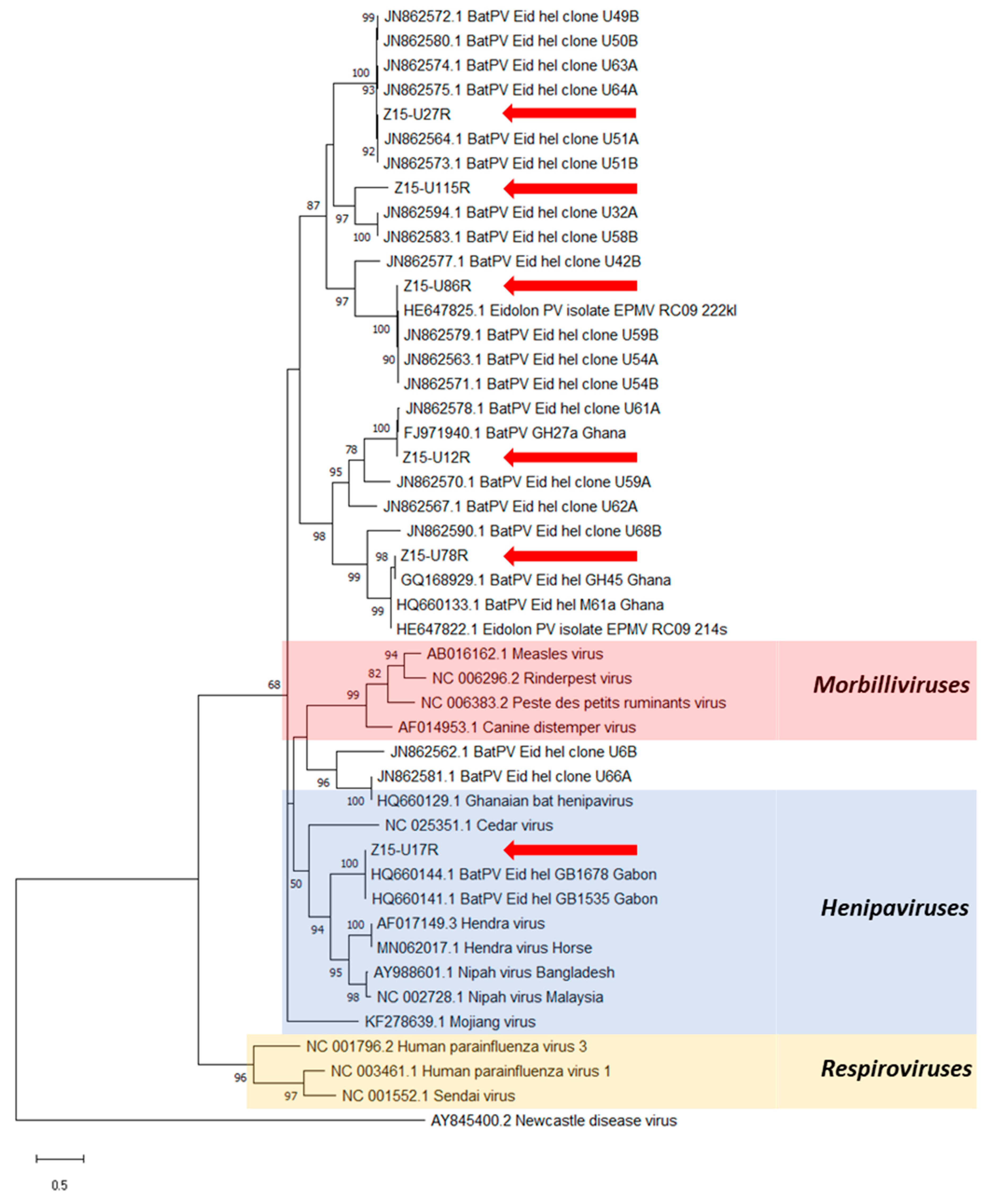
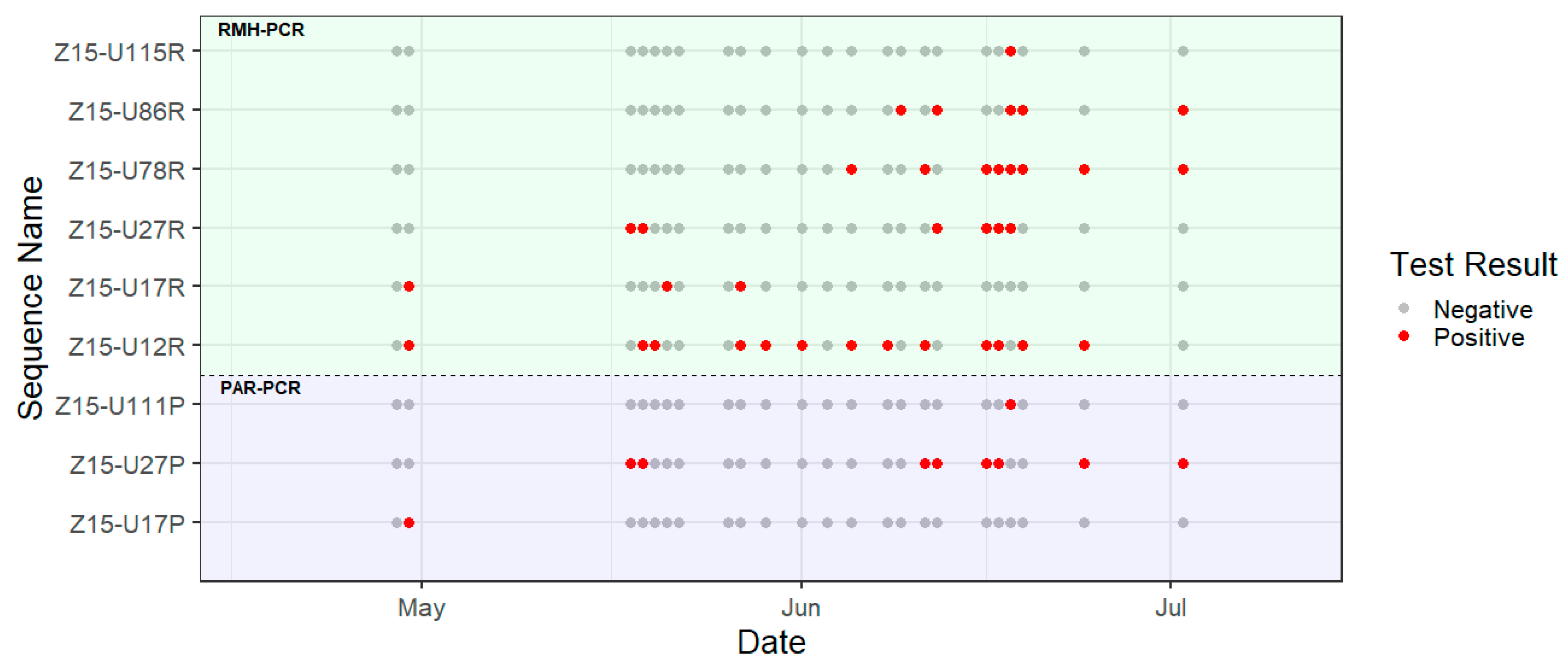
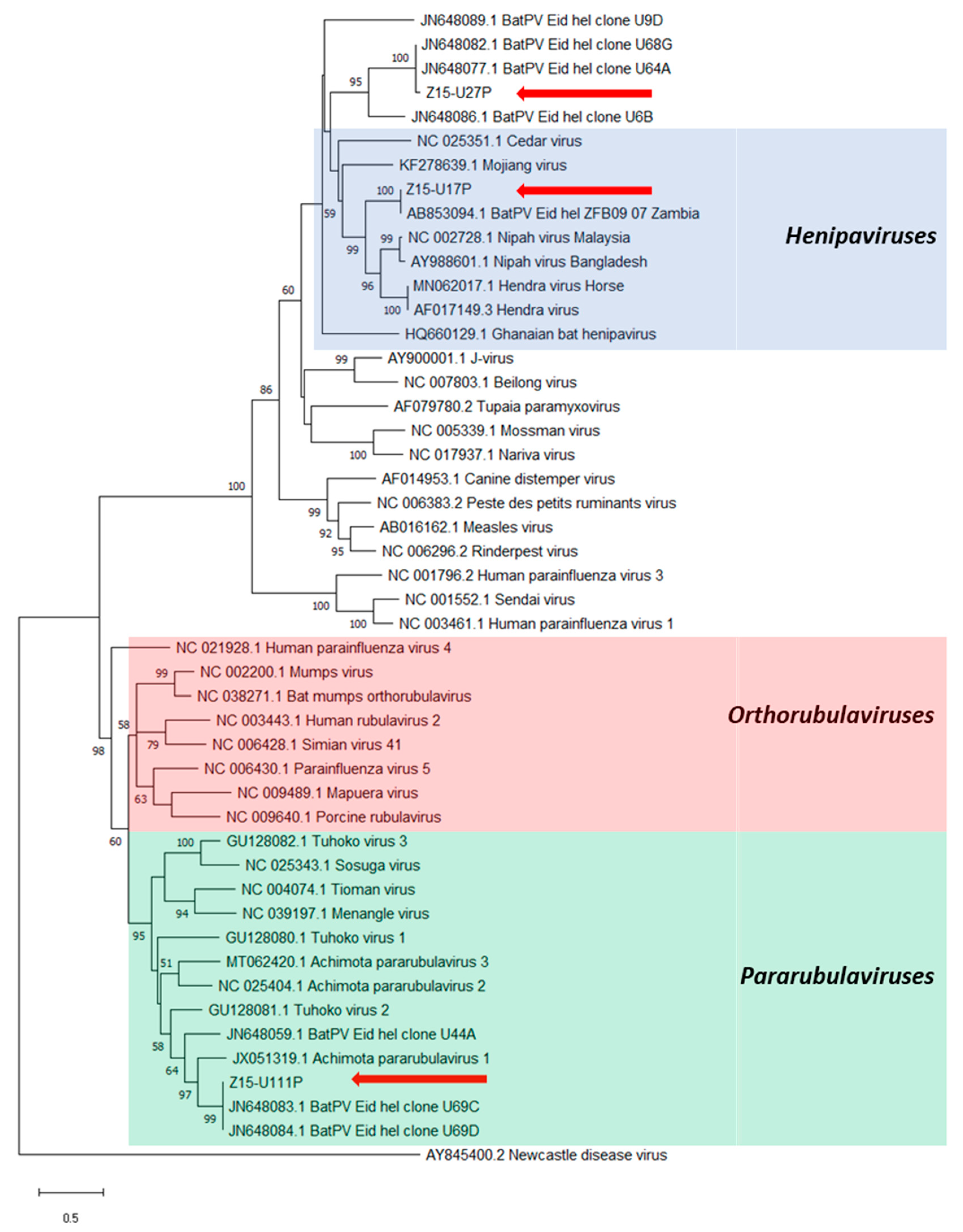
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Muleya, W.; Sasaki, M.; Orba, Y.; Ishii, A.; Thomas, Y.; Nakagawa, E.; Ogawa, H.; Hang’ombe, B.; Namangala, B.; Mweene, A.; et al. Molecular epidemiology of paramyxoviruses in Frugivorous Eidolon helvum bats in Zambia. J. Vet. Med. Sci. 2014, 76, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suu-Ire, R.; Begeman, L.; Banyard, A.C.; Breed, A.C.; Drosten, C.; Eggerbauer, E.; Freuling, C.M.; Gibson, L.; Goharriz, H.; Horton, D.L.; et al. Pathogenesis of bat rabies in a natural reservoir: Comparative susceptibility of the straw-colored fruit bat (Eidolon helvum) to three strains of Lagos bat virus. PLoS Negl. Trop. Dis. 2018, 12, e0006311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luis, A.D.; Hayman, D.T.S.; O’shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.C.; Mills, J.N.; Willis, C.K.R.; Timonin, M.E.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. 2013, 280, 20122753. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.Y. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef]
- Luis, A.D.; O’Shea, T.J.; Hayman, D.T.S.; Wood, J.L.N.; Cunningham, A.A.; Gilbert, A.T.; Mills, J.N.; Webb, C.T. Network analysis of host-virus communities in bats and rodents reveals determinants of cross-species transmission. Ecol. Lett. 2015, 18, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, T.J.; Cryan, P.M.; Cunningham, A.A.; Fooks, A.R.; Hayman, D.T.S.; Luis, A.D.; Peel, A.J.; Plowright, R.K.; Wood, J.L.N. Bat flight and zoonotic viruses. Emerg. Infect. Dis. 2014, 20, 741–745. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.; Rogers, R.; Selvey, L.; Selleck, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Hooper, P.; Westbury, H. A novel morbillivirus pneumonia of horses and its transmission to humans. Emerg. Infect. Dis. 1995, 1, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Broos, A.; de Jong, C.; Zeddeman, A.; Smith, C.; Smith, G.; Moore, F.; Barr, J.; Crameri, G.; Marsh, G.; et al. Identifying hendra virus diversity in pteropid bats. PLoS ONE 2011, 6, e25275. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Hsu, V.P.; Hossain, M.J.; Parashar, U.D.; Ali, M.M.; Ksiazek, T.G.; Kuzmin, I.; Niezgoda, M.; Rupprecht, C.; Bresee, J.; Breiman, R.F. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis. 2004, 10, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Luby, S.P.; Rahman, M.; Hossain, M.J.; Blum, L.S.; Husain, M.M.; Gurley, E.; Khan, R.; Ahmed, B.N.; Rahman, S.; Nahar, N.; et al. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 2006, 12, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Rota, P.A.; Rollin, P.E. A review of Nipah and Hendra viruses with an historical aside. Virus Res. 2011, 162, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.S.; Todd, S.; Marsh, G.; Fernandez-Loras, A.; Suu-Ire, R.; Wood, J.L.N.; Wang, L.F.; Murcia, P.R.; Cunningham, A.A. Co-circulation of diverse paramyxoviruses in an urban African fruit bat population. J. Gen. Virol. 2012, 93, 850–856. [Google Scholar] [CrossRef]
- Mortlock, M.; Kuzmin, I.V.; Weyer, J.; Gilbert, A.T.; Agwanda, B.; Rupprecht, C.E.; Nel, L.H.; Kearney, T.; Malekani, J.M.; Markotter, W. Novel paramyxoviruses in bats from Sub-Saharan Africa, 2007–2012. Emerg. Infect. Dis. 2015, 21, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.; Smith, C.; Smith, I.; De Jong, C.; Todd, S.; Melville, D.; Broos, A.; Crameri, S.; Haining, J.; Marsh, G.; et al. Isolation of multiple novel paramyxoviruses from pteropid bat urine. J. Gen. Virol. 2015, 96, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Gloza-Rausch, F.; Seebens, A.; Annan, A.; Ipsen, A.; Kruppa, T.; Müller, M.A.; Kalko, E.K.V.; Adu-Sarkodie, Y.; et al. Henipavirus RNA in African bats. PLoS ONE 2009, 4, e6367. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.S.; Todd, S.; Marsh, G.A.; Crameri, G.; Barr, J.; Kamins, A.O.; Peel, A.J.; Yu, M.; Hayman, D.T.S.; Nadjm, B.; et al. Novel, Potentially Zoonotic Paramyxoviruses from the African Straw-Colored Fruit Bat Eidolon helvum. J. Virol. 2013, 87, 1348–1358. [Google Scholar] [CrossRef] [Green Version]
- Hayman, D.T.S.; Suu-Ire, R.; Breed, A.C.; McEachern, J.A.; Wang, L.; Wood, J.L.N.; Cunningham, A.A. Evidence of henipavirus infection in West African fruit bats. PLoS ONE 2008, 3, e2739. [Google Scholar] [CrossRef]
- Voigt, K.; Hoffmann, M.; Drexler, J.F.; Müller, M.A.; Drosten, C.; Herrler, G.; Krüger, N. Fusogenicity of the Ghana Virus (Henipavirus: Ghanaian bat henipavirus) Fusion Protein is Controlled by the Cytoplasmic Domain of the Attachment Glycoprotein. Viruses 2019, 11, 800. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; Lebreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for henipavirus spillover into human populations in Africa. Nat. Commun. 2014, 5, 5342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philbey, A.W.; Kirkland, P.D.; Ross, A.D.; Davis, R.J.; Gleeson, A.B.; Love, R.J.; Daniels, P.W.; Gould, A.R.; Hyatt, A.D. An apparently new virus (family Paramyxoviridae) infectious for pigs, humans, and fruit bats. Emerg. Infect. Dis. 1998, 4, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.B.; Wang, L.F.; Lam, S.K.; Crameri, G.; Yu, M.; Wise, T.; Boyle, D.; Hyatt, A.D.; Eaton, B.T. Tioman virus, a novel paramyxovirus isolated from fruit bats in Malaysia. Virology 2001, 283, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Woo, P.C.Y.; Wong, B.H.L.; Wong, A.Y.P.; Tsoi, H.W.; Wang, M.; Lee, P.; Xu, H.; Poon, R.W.S.; Guo, R.; et al. Identification and complete genome analysis of three novel paramyxoviruses, Tuhoko virus 1, 2 and 3, in fruit bats from China. Virology 2010, 404, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.S.; Tachedjian, M.; Barr, J.; Marsh, G.A.; Todd, S.; Crameri, G.; Crameri, S.; Smith, I.; Holmes, C.E.G.; Suu-Ire, R.; et al. Achimota Pararubulavirus 3: A New Bat-Derived Paramyxovirus of the Genus Pararubulavirus. Viruses 2020, 12, 1236. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.; Todd, S.; Crameri, G.; Foord, A.; Marsh, G.; Frazer, L.; Payne, J.; Harper, J.; Baker, K.S.; Cunningham, A.A.; et al. Animal infection studies of two recently discovered African bat paramyxoviruses, Achimota 1 and Achimota 2. Sci. Rep. 2018, 8, 12744. [Google Scholar] [CrossRef] [Green Version]
- Mickleburgh, S.; Waylen, K.; Racey, P. Bats as bushmeat: A global review. ORYX 2009, 43, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Peel, A.J.; Sargan, D.R.; Baker, K.S.; Hayman, D.T.S.; Barr, J.A.; Crameri, G.; Suu-Ire, R.; Broder, C.C.; Lembo, T.; Wang, L.F.; et al. Continent-wide panmixia of an African fruit bat facilitates transmission of potentially zoonotic viruses. Nat. Commun. 2013, 4, 2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, K.S.; Suu-Ire, R.; Barr, J.; Hayman, D.T.S.; Broder, C.C.; Horton, D.L.; Durrant, C.; Murcia, P.R.; Cunningham, A.A.; Wood, J.L.N. Viral antibody dynamics in a chiropteran host. J. Anim. Ecol. 2014, 83, 415–428. [Google Scholar] [CrossRef]
- Peel, A.J.; McKinley, T.J.; Baker, K.S.; Barr, J.A.; Crameri, G.; Hayman, D.T.S.; Feng, Y.R.; Broder, C.C.; Wang, L.F.; Cunningham, A.A.; et al. Use of cross-reactive serological assays for detecting novel pathogens in wildlife: Assessing an appropriate cutoff for henipavirus assays in African bats. J. Virol. Methods 2013, 193, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Glennon, E.E.; Becker, D.J.; Peel, A.J.; Garnier, R.; Suu-Ire, R.D.; Gibson, L.; Hayman, D.T.S.; Wood, J.L.N.; Cunningham, A.A.; Plowright, R.K.; et al. What is stirring in the reservoir? Modelling mechanisms of henipavirus circulation in fruit bat hosts. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190021. [Google Scholar] [CrossRef] [Green Version]
- Edson, D.; Field, H.; McMichael, L.; Vidgen, M.; Goldspink, L.; Broos, A.; Melville, D.; Kristoffersen, J.; De Jong, C.; McLaughlin, A.; et al. Routes of hendra virus excretion in naturally-infected flying-foxes: Implications for viral transmission and spillover risk. PLoS ONE 2015, 10, e0140670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Chern, S.W.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- NCBI Nucleotide BLAST: Search Nucleotide Databases Using a Nucleotide Query. Basic Local Alignment Search Tool. 2015. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 15 June 2021).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.; Azouri, D.; Pupko, T.; Mayrose, I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Nowak, K.; Fahr, J.; Wibbelt, G.; Mombouli, J.V.; Parra, H.J.; Wolfe, N.D.; Schneider, B.S.; Leendertz, F.H. Henipavirus-related sequences in fruit bat bushmeat, Republic of Congo. Emerg. Infect. Dis. 2012, 18, 1536–1537. [Google Scholar] [CrossRef]
- Pomeroy, L.W.; Bjørnstad, O.N.; Holmes, E.C. The evolutionary and epidemiological dynamics of the paramyxoviridae. J. Mol. Evol. 2008, 66, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, M.S. Measles Periodicity and Community Size. J. R. Stat. Soc. Ser. A 1957, 120, 48–60. [Google Scholar] [CrossRef]
- Peel, A.J.; Baker, K.S.; Crameri, G.; Barr, J.A.; Hayman, D.T.S.; Wright, E.; Broder, C.C.; Fernández-Loras, A.; Fooks, A.R.; Wang, L.F.; et al. Henipavirus neutralising antibodies in an isolated island population of African fruit bats. PLoS ONE 2012, 7, e30346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.A.; Hassan, S.S.; Olival, K.J.; Mohamed, M.; Chang, L.Y.; Hassan, L.; Saad, N.M.; Shohaimi, S.A.; Mamat, Z.C.; Naim, M.S.; et al. Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerg. Infect. Dis. 2010, 16, 1990–1993. [Google Scholar] [CrossRef]
- Plowright, R.K.; Field, H.E.; Smith, C.; Divljan, A.; Palmer, C.; Tabor, G.; Daszak, P.; Foley, J.E. Reproduction and nutritional stress are risk factors for Hendra virus infection in little red flying foxes (Pteropus scapulatus). Proc. R. Soc. B Biol. Sci. 2008, 275, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Peel, A.J.; Baker, K.S.; Hayman, D.T.S.; Broder, C.C.; Cunningham, A.A.; Fooks, A.R.; Garnier, R.; Wood, J.L.N.; Restif, O. Support for viral persistence in bats from age-specific serology and models of maternal immunity. Sci. Rep. 2018, 3859, 1–11. [Google Scholar] [CrossRef]
- Kamins, A.O.; Restif, O.; Ntiamoa-Baidu, Y.; Suu-Ire, R.; Hayman, D.T.S.; Cunningham, A.A.; Wood, J.L.N.; Rowcliffe, J.M. Uncovering the fruit bat bushmeat commodity chain and the true extent of fruit bat hunting in Ghana, West Africa. Biol. Conserv. 2011, 144, 3000–3008. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | No. of Samples Tested | No. of PCR Positive Amplicons | |
|---|---|---|---|
| PAR-PCR | RMH-PCR | ||
| 29/04/2015 | 10 | 0 | 0 |
| 30/04/2015 | 15 | 1 | 7 |
| 18/05/2015 | 5 | 4 | 4 |
| 19/05/2015 | 5 | 1 | 3 |
| 20/05/2015 | 5 | 0 | 1 |
| 21/05/2015 | 5 | 0 | 1 |
| 22/05/2015 | 5 | 0 | 0 |
| 26/05/2015 | 3 | 0 | 0 |
| 27/05/2015 | 5 | 0 | 4 |
| 29/05/2015 | 5 | 0 | 1 |
| 01/06/2015 | 5 | 0 | 3 |
| 03/06/2015 | 5 | 0 | 0 |
| 05/06/2015 | 5 | 0 | 4 |
| 08/06/2015 | 5 | 0 | 5 |
| 09/06/2015 | 5 | 0 | 4 |
| 11/06/2015 | 5 | 1 | 2 |
| 12/06/2015 | 5 | 1 | 2 |
| 16/06/2015 | 5 | 4 | 4 |
| 17/06/2015 | 5 | 2 | 5 |
| 18/06/2015 | 5 | 1 | 5 |
| 19/06/2015 | 5 | 0 | 5 |
| 24/06/2015 | 5 | 1 | 4 |
| 02/07/2015 | 5 | 1 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibson, L.; Ribas, M.P.; Kemp, J.; Restif, O.; Suu-Ire, R.D.; Wood, J.L.N.; Cunningham, A.A. Persistence of Multiple Paramyxoviruses in a Closed Captive Colony of Fruit Bats (Eidolon helvum). Viruses 2021, 13, 1659. https://doi.org/10.3390/v13081659
Gibson L, Ribas MP, Kemp J, Restif O, Suu-Ire RD, Wood JLN, Cunningham AA. Persistence of Multiple Paramyxoviruses in a Closed Captive Colony of Fruit Bats (Eidolon helvum). Viruses. 2021; 13(8):1659. https://doi.org/10.3390/v13081659
Chicago/Turabian StyleGibson, Louise, Maria Puig Ribas, James Kemp, Olivier Restif, Richard D. Suu-Ire, James L. N. Wood, and Andrew A. Cunningham. 2021. "Persistence of Multiple Paramyxoviruses in a Closed Captive Colony of Fruit Bats (Eidolon helvum)" Viruses 13, no. 8: 1659. https://doi.org/10.3390/v13081659
APA StyleGibson, L., Ribas, M. P., Kemp, J., Restif, O., Suu-Ire, R. D., Wood, J. L. N., & Cunningham, A. A. (2021). Persistence of Multiple Paramyxoviruses in a Closed Captive Colony of Fruit Bats (Eidolon helvum). Viruses, 13(8), 1659. https://doi.org/10.3390/v13081659