
Islatravir Is Not Expected to Be a Victim or Perpetrator of Drug-Drug Interactions via Major Drug-Metabolizing Enzymes or Transporters

Abstract
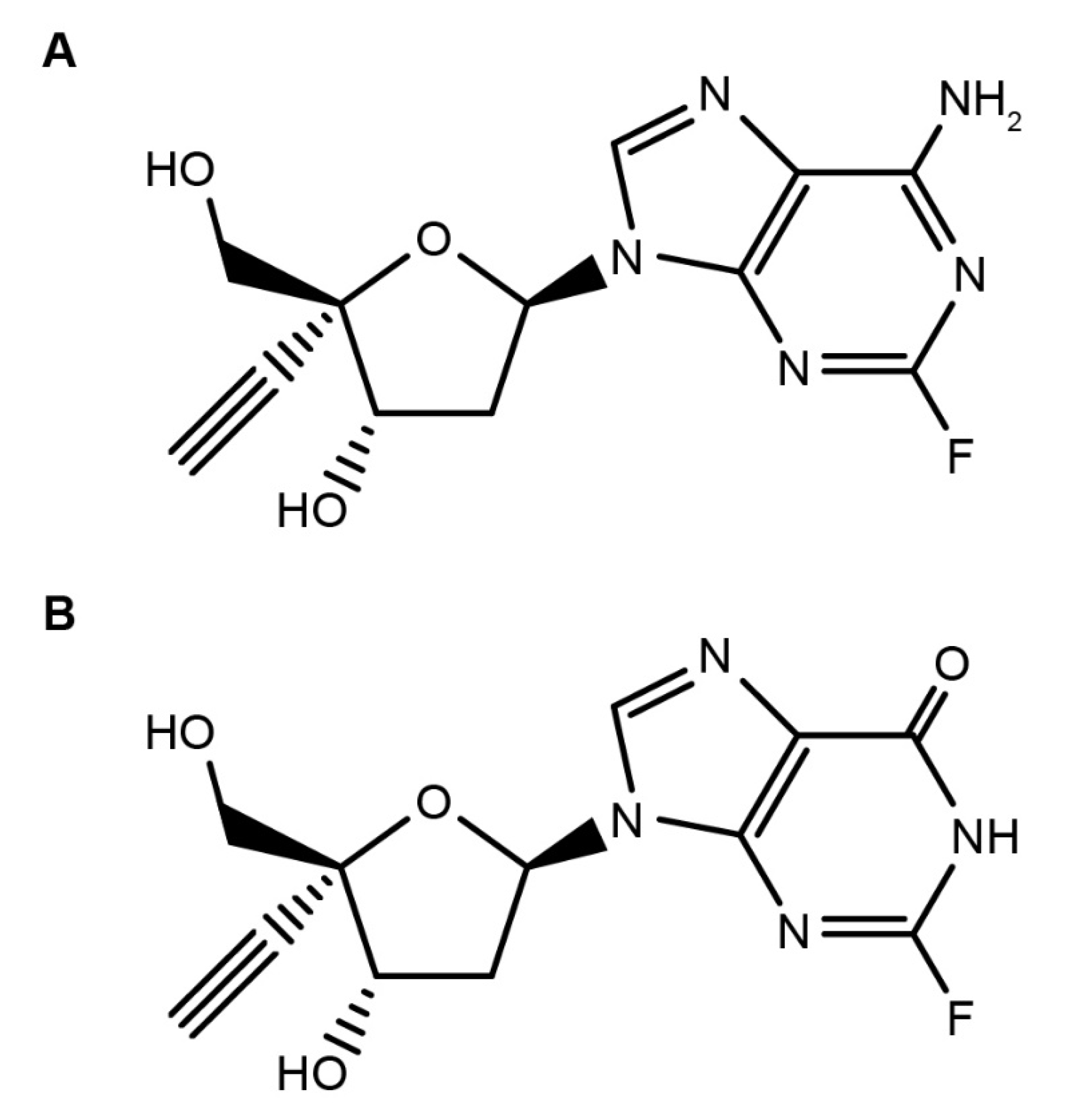
1. Introduction
2. Materials and Methods
2.1. Islatravir Distribution in Plasma
2.2. Characterization of Islatravir Metabolism in Intestinal S9 and Metabolism by Human Adenosine Deaminase
2.3. Characterization of Renal Clearance in Animal Models
2.4. Interaction of Islatravir with Drug-Metabolizing Enzymes: CYP Isoforms and UGT1A1
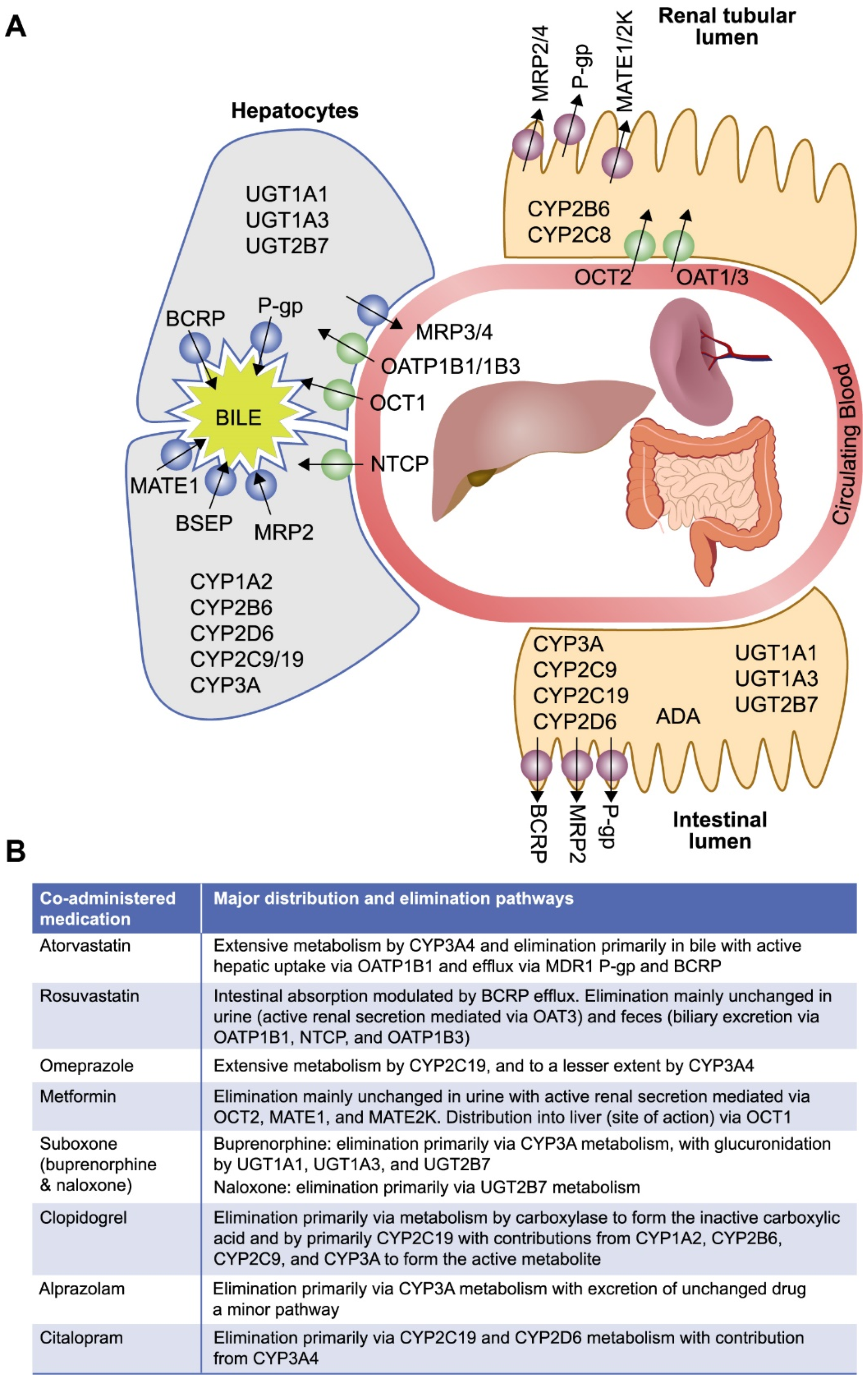
2.5. Evaluation of Islatravir as a Perpetrator of Drug–Drug Interactions via Transporters
2.6. Evaluation of Islatravir as a Victim of Drug–Drug Interactions via Transporters
3. Results
3.1. Islatravir Exhibited Low Plasma Protein Binding
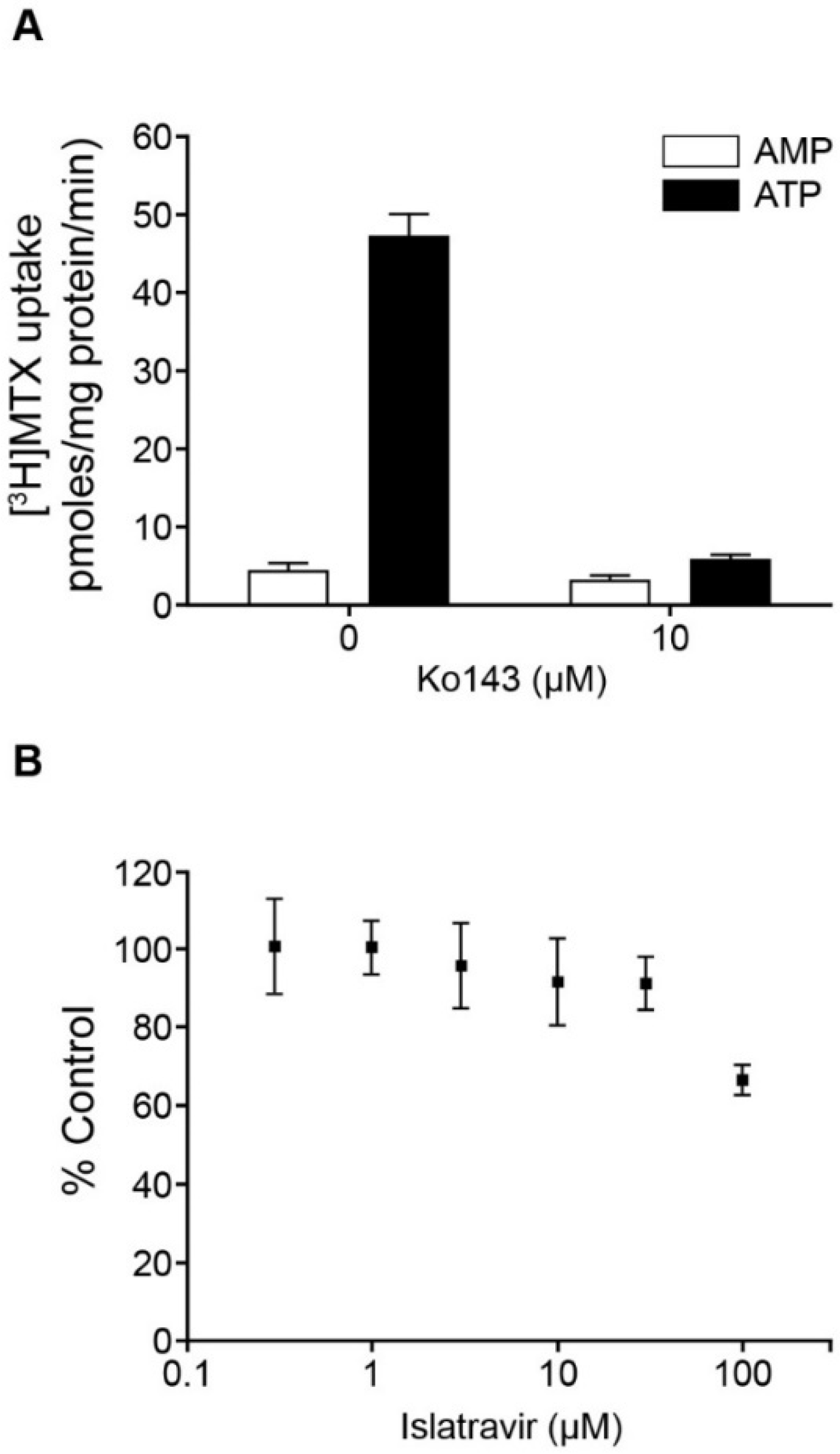
3.2. Islatravir Was Metabolized by Adenosine Deaminase
3.3. Islatravir Was Partially Cleared via Renal Excretion in Nonclinical Species
3.4. Islatravir Did Not Inhibit or Induce Major Drug-Metabolizing Enzymes
3.5. Islatravir Did Not Inhibit Major Hepatic Transporters at Clinically Relevant Concentrations
3.6. Islatravir Did Not Inhibit Major Renal Transporters at Clinically Relevant Concentrations
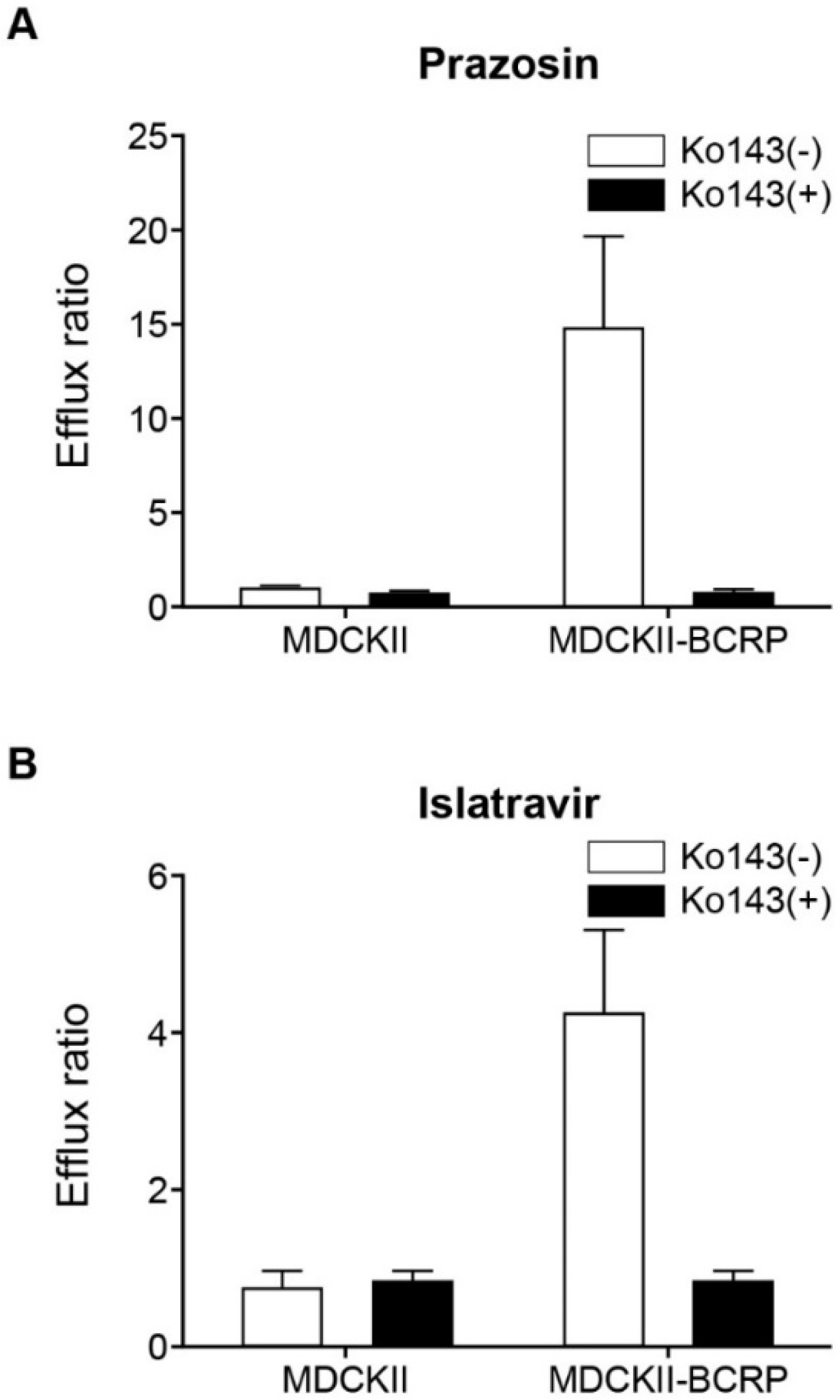
3.7. Islatravir Did Not Inhibit MDR1 P-gp and BCRP at Clinically Relevant Concentrations
3.8. Islatravir Was Not a Substrate of Major Renal Transporters
3.9. Islatravir Was a Substrate of BCRP, but Not MDR1 P-gp
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antiretroviral Therapy Cohort Collaboration. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: A collaborative analysis of 14 cohort studies. Lancet 2008, 372, 293–299. [Google Scholar] [CrossRef]
- Marcus, J.L.; Leyden, W.A.; Alexeeff, S.E.; Anderson, A.N.; Hechter, R.C.; Hu, H.; Lam, J.O.; Towner, W.J.; Yuan, Q.; Horberg, M.A.; et al. Comparison of Overall and Comorbidity-Free Life Expectancy between Insured Adults with and without HIV Infection, 2000–2016. JAMA Netw. Open 2020, 3, e207954. [Google Scholar] [CrossRef]
- UNAIDS. UNAIDS Fact Sheet—Global AIDS Update 2019. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 22 July 2021).
- Wada, N.; Jacobson, L.P.; Cohen, M.; French, A.; Phair, J.; Munoz, A. Cause-specific life expectancies after 35 years of age for human immunodeficiency syndrome-infected and human immunodeficiency syndrome-negative individuals followed simultaneously in long-term cohort studies, 1984–2008. Am. J. Epidemiol. 2013, 177, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Eisinger, R.W.; Fauci, A.S. Comorbidities in Persons with HIV: The Lingering Challenge. JAMA 2019, 323, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.; Brinkman, K.; Geerlings, S.; Smit, C.; Thyagarajan, K.; Sighem, A.; de Wolf, F.; Hallett, T.B.; Cohort, A.O. Future challenges for clinical care of an ageing population infected with HIV: A modelling study. Lancet Infect. Dis. 2015, 15, 810–818. [Google Scholar] [CrossRef]
- Friedman, E.E.; Duffus, W.A. Chronic health conditions in Medicare beneficiaries 65 years old, and older with HIV infection. AIDS 2016, 30, 2529–2536. [Google Scholar] [CrossRef]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef]
- Back, D.; Marzolini, C. The challenge of HIV treatment in an era of polypharmacy. J. Int. AIDS Soc. 2020, 23, e25449. [Google Scholar] [CrossRef]
- Escota, G.V.; O’Halloran, J.A.; Powderly, W.G.; Presti, R.M. Understanding mechanisms to promote successful aging in persons living with HIV. Int. J. Infect. Dis. 2018, 66, 56–64. [Google Scholar] [CrossRef]
- Gallant, J.; Hsue, P.Y.; Shreay, S.; Meyer, N. Comorbidities among US patients with prevalent HIV infection-a trend analysis. J. Infect. Dis. 2017, 216, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.; Steinman, M.A.; McNicholl, I.R.; Valcour, V. Polypharmacy, drug-drug interactions, and potentially inappropriate medications in older adults with human immunodeficiency virus infection. J. Am. Geriatr. Soc. 2014, 62, 447–453. [Google Scholar] [CrossRef]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical studies on drug-drug interactions involving metabolism and transport: Methodology, pitfalls, and interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 22 July 2021).
- US Food and Drug Administration Center for Drug Evaluation and Research. Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 22 July 2021).
- PMDA Ministry of Labor and Welfare Guideline on Drug Interaction for Drug Development and Appropriate Provision of Information, Notification No.0723-4, Pharmaceutical Evaluation Division, Pharmaceuticals Safety and Environmental Health Bureau, Japan. 23 July 2018. Available online: https://www.pmda.go.jp/files/000228122.pdf (accessed on 22 July 2021).
- Markowitz, M.; Grobler, J.A. Islatravir for the treatment and prevention of infection with the human immunodeficiency virus type 1. Curr. Opin. HIV AIDS 2020, 15, 27–32. [Google Scholar] [CrossRef]
- Markowitz, M.; Sarafianos, S.G. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine, MK-8591: A novel HIV-1 reverse transcriptase translocation inhibitor. Curr. Opin. HIV AIDS 2018, 13, 294–299. [Google Scholar] [CrossRef]
- Kawamoto, A.; Kodama, E.; Sarafianos, S.G.; Sakagami, Y.; Kohgo, S.; Kitano, K.; Ashida, N.; Iwai, Y.; Hayakawa, H.; Nakata, H.; et al. 2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Huber, A.D.; Ryan, E.M.; Ong, Y.T.; Leslie, M.D.; Matzek, K.B.; Singh, K.; Marchand, B.; Hagedorn, A.N.; Kirby, K.A.; et al. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J. Biol. Chem. 2014, 289, 24533–24548. [Google Scholar] [CrossRef] [PubMed]
- Salie, Z.L.; Kirby, K.A.; Michailidis, E.; Marchand, B.; Singh, K.; Rohan, L.C.; Kodama, E.N.; Mitsuya, H.; Parniak, M.A.; Sarafianos, S.G. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. USA 2016, 113, 9274–9279. [Google Scholar] [CrossRef] [PubMed]
- DHHS. Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. 2019. Available online: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultandAdolescentGL.pdf (accessed on 22 July 2021).
- Grobler, J.; Lai, M.T.; Barrett, S.E.; Gindy, M.; Fillgrove, K.; Ankrom, W.; Sandra, W.; Friedman, E.; Iwamoto, M.; Hazuda, D.J. Long-acting oral and parenteral dosing of MK-8591 for HIV treatment or prophylaxis. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI), Boston, MA, USA, 22–25 February 2016. [Google Scholar]
- Michailidis, E.; Marchand, B.; Kodama, E.N.; Singh, K.; Matsuoka, M.; Kirby, K.A.; Ryan, E.M.; Sawani, A.M.; Nagy, E.; Ashida, N.; et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-Ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, C.A.; Galkina, S.A.; Joshi, P.; Kosikova, G.; Moreno, M.E.; Rivera, J.M.; Sloan, B.; Reeve, A.B.; Sarafianos, S.G.; Murphey-Corb, M.; et al. Oral administration of the nucleoside EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob. Agents Chemother. 2015, 59, 4190–4198. [Google Scholar] [CrossRef] [PubMed]
- Schurmann, D.; Rudd, D.J.; Zhang, S.; De Lepeleire, I.; Robberechts, M.; Friedman, E.; Keicher, C.; Huser, A.; Hofmann, J.; Grobler, J.A.; et al. Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK-8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single-dose administration to treatment-naive adults infected with HIV-1: An open-label, phase 1b, consecutive-panel trial. Lancet HIV 2020, 7, e164–e172. [Google Scholar] [CrossRef]
- Matthews, R.P.; Jackson Rudd, D.; Levine, V.; Zhang, S.; Sterling, L.; Grobler, J.A.; Vargo, R.; Stoch, S.A.; Iwamoto, M. Multiple daily doses of MK-8591 as low as 0.25 mg are expected to suppress HIV. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI), Boston, MA, USA, 4–7 March 2018. [Google Scholar]
- Clinicaltrials.gov. Safety and Pharmacokinetics of Oral Islatravir (MK-8591) Once Monthly in Participants at Low Risk of Human Immunodeficiency Virus 1 (HIV-1) Infection (MK-8591-016) (NCT04003103). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04003103 (accessed on 22 July 2021).
- Hillier, S.; Bekker, L.G.; Badal-Faesen, S.; Hendrix, C.W.; Riddler, S.A.; Rasmussen, S.; Schwartz, H.; Nair, G.; Lombaard, J.H.; Caraco, Y.; et al. Trial design, enrollment status, demographics, and pharmacokinetic (PK) data from a blinded interim analysis from a phase 2a trial of islatravir once monthly (QM) for HIV pre-exposure prophylaxis (PrEP). In Proceedings of the Conference on HIV Research for Prevention (HIVR4P), Virtual, 27–28 January 2021 and 3–4 February 2021. [Google Scholar]
- US Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhbitors and Inducers. 2020. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table2-1 (accessed on 22 July 2021).
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharm. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport--an update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Pfeifer, N.D.; Hardwick, R.N.; Brouwer, K.L. Role of hepatic efflux transporters in regulating systemic and hepatocyte exposure to xenobiotics. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 509–535. [Google Scholar] [CrossRef] [PubMed]
- Kellick, K.A.; Bottorff, M.; Toth, P.P. The National Lipid Association’s Safety Task F. A clinician’s guide to statin drug-drug interactions. J. Clin. Lipidol. 2014, 8, S30–S46. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.E.; Zolk, O.; Fromm, M.F.; Kurkinen, K.J.; Neuvonen, P.J.; Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2009, 86, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Elsby, R.; Hilgendorf, C.; Fenner, K. Understanding the critical disposition pathways of statins to assess drug-drug interaction risk during drug development: It’s not just about OATP1B1. Clin. Pharmacol. Ther. 2012, 92, 584–598. [Google Scholar] [CrossRef]
- Shirasaka, Y.; Sager, J.E.; Lutz, J.D.; Davis, C.; Isoherranen, N. Inhibition of CYP2C19 and CYP3A4 by omeprazole metabolites and their contribution to drug-drug interactions. Drug Metab. Dispos. 2013, 41, 1414–1424. [Google Scholar] [CrossRef]
- Stolbach, A.; Paziana, K.; Heverling, H.; Pham, P. A review of the toxicity of HIV medications II: Interactions with drugs and complementary and alternative medicine products. J. Med. Toxicol. 2015, 11, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Bottiger, Y.; Tybring, G.; Gotharson, E.; Bertilsson, L. Inhibition of the sulfoxidation of omeprazole by ketoconazole in poor and extensive metabolizers of S-mephenytoin. Clin. Pharmacol. Ther. 1997, 62, 384–391. [Google Scholar] [CrossRef]
- May, M.; Schindler, C. Clinically and pharmacologically relevant interactions of antidiabetic drugs. Ther. Adv. Endocrinol. Metab. 2016, 7, 69–83. [Google Scholar] [CrossRef]
- Wiebe, S.T.; Giessmann, T.; Hohl, K.; Schmidt-Gerets, S.; Hauel, E.; Jambrecina, A.; Bader, K.; Ishiguro, N.; Taub, M.E.; Sharma, A.; et al. Validation of a drug transporter probe cocktail using the prototypical inhibitors rifampin, probenecid, verapamil, and cimetidine. Clin. Pharm. 2020, 59, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharm. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Moody, D.E.; Fang, W.B.; Lin, S.N.; Weyant, D.M.; Strom, S.C.; Omiecinski, C.J. Effect of rifampin and nelfinavir on the metabolism of methadone and buprenorphine in primary cultures of human hepatocytes. Drug Metab. Dispos. 2009, 37, 2323–2329. [Google Scholar] [CrossRef]
- Coffman, B.L.; King, C.D.; Rios, G.R.; Tephly, T.R. The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y(268) and UGT2B7H(268). Drug Metab. Dispos. 1998, 26, 73–77. [Google Scholar]
- Cheng, Z.; Rios, G.R.; King, C.D.; Coffman, B.L.; Green, M.D.; Mojarrabi, B.; Mackenzie, P.I.; Tephly, T.R. Glucuronidation of catechol estrogens by expressed human UDP-glucuronosyltransferases (UGTs) 1A1, 1A3, and 2B7. Toxicol. Sci. 1998, 45, 52–57. [Google Scholar] [CrossRef][Green Version]
- Papathanasiou, T.; Springborg, A.D.; Kongstad, K.T.; Staerk, D.; Moller, K.; Taylor, B.K.; Lund, T.M.; Werner, M.U. High-dose naloxone, an experimental tool uncovering latent sensitisation: Pharmacokinetics in humans. Br. J. Anaesth. 2019, 123, e204–e214. [Google Scholar] [CrossRef]
- Jiang, X.L.; Samant, S.; Lesko, L.J.; Schmidt, S. Clinical pharmacokinetics and pharmacodynamics of clopidogrel. Clin. Pharm. 2015, 54, 147–166. [Google Scholar] [CrossRef]
- Mistry, S.D.; Trivedi, H.R.; Parmar, D.M.; Dalvi, P.S.; Jiyo, C. Impact of proton pump inhibitors on efficacy of clopidogrel: Review of evidence. Indian J. Pharmacol. 2011, 43, 183–186. [Google Scholar] [CrossRef]
- Bonello, L.; Tantry, U.S.; Marcucci, R.; Blindt, R.; Angiolillo, D.J.; Becker, R.; Bhatt, D.L.; Cattaneo, M.; Collet, J.P.; Cuisset, T.; et al. Consensus and future directions on the definition of high on-treatment platelet reactivity to adenosine diphosphate. J. Am. Coll Cardiol. 2010, 56, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, K.; Greenblatt, D.J.; von Moltke, L.L.; Shader, R.I. Alprazolam is another substrate for human cytochrome P450-3A isoforms. J. Clin. Psychopharmacol. 1998, 18, 256. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef]
- Baumann, P. Pharmacology and pharmacokinetics of citalopram and other SSRIs. Int. Clin. Psychopharmacol. 1996, 11 (Suppl. 1), 5–11. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Chiba, K.; Yagi, T.; Shimada, N.; Taniguchi, T.; Horie, T.; Tani, M.; Yamamoto, T.; Ishizaki, T.; Kuroiwa, Y. Identification of cytochrome P450 isoforms involved in citalopram N-demethylation by human liver microsomes. J. Pharmacol. Exp. Ther. 1997, 280, 927–933. [Google Scholar] [PubMed]
- Sanchez, R.I.; Fillgrove, K.L.; Yee, K.L.; Liang, Y.; Lu, B.; Tatavarti, A.; Liu, R.; Anderson, M.S.; Behm, M.O.; Fan, L.; et al. Characterisation of the absorption, distribution, metabolism, excretion and mass balance of doravirine, a non-nucleoside reverse transcriptase inhibitor in humans. Xenobiotica 2019, 49, 422–432. [Google Scholar] [CrossRef]
- Bleasby, K.; Fillgrove, K.L.; Houle, R.; Lu, B.; Palamanda, J.; Newton, D.J.; Lin, M.; Chan, G.H.; Sanchez, R.I. In vitro evaluation of the drug interaction potential of doravirine. Antimicrob. Agents Chemother. 2019, 63, e02492-18. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, O.A.; Shebley, M.; Palamanda, J.; Sinz, M.W.; Ramsden, D.; Einolf, H.J.; Chen, L.; Wang, H. Evaluation of CYP2B6 induction and prediction of clinical drug-drug interactions: Considerations from the IQ Consortium Induction Working Group-an industry perspective. Drug Metab. Dispos. 2016, 44, 1720–1730. [Google Scholar] [CrossRef]
- Chu, X.; Cai, X.; Cui, D.; Tang, C.; Ghosal, A.; Chan, G.; Green, M.; Kuo, Y.; Liang, Y.; Maciolek, C.; et al. In vitro assessment of drug-drug interaction potential of boceprevir associated with drug metabolizing enzymes and transporters. Drug Metab. Dispos. 2013, 41, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Rizk, M.; Houle, R.; Chan, G.; Hafey, M.; Rhee, E.; Chu, X. Raltegravir has a low propensity to cause clinical drug interactions through inhibition of major drug transporters: An in vitro evaluation. Antimicrob. Agents Chemother. 2014, 58, 1294–1301. [Google Scholar] [CrossRef]
- Chu, X.; Bleasby, K.; Yabut, J.; Cai, X.; Chan, G.; Hafey, M.; Xu, S.; Bergman, A.; Braun, M.; Dean, D.; et al. Transport of the dipeptidyl peptidase-4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P-glycoprotein. J. Pharmacol. Exp. Ther. 2007, 321, 673–683. [Google Scholar] [CrossRef]
- The Human Protein Atlas. ADA. 2020. Available online: https://www.proteinatlas.org/ENSG00000196839-ADA/tissue (accessed on 22 July 2021).
- Kirby, K.A.; Michailidis, E.; Fetterly, T.L.; Steinbach, M.A.; Singh, K.; Marchand, B.; Leslie, M.D.; Hagedorn, A.N.; Kodama, E.N.; Marquez, V.E.; et al. Effects of substitutions at the 4′ and 2 positions on the bioactivity of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 2013, 57, 6254–6264. [Google Scholar] [CrossRef]
- Austin, R.; Barton, P.; Cockroft, S.; Wenlock, M.; Riley, R. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos. 2002, 30, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, E. Clinically important pharmacokinetic drug-drug interactions: Role of cytochrome P450 enzymes. J. Clin. Pharm. Ther. 1998, 23, 403–416. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Low, Y.; Setia, S.; Lima, G. Drug-drug interactions involving antidepressants: Focus on desvenlafaxine. Neuropsychiatr. Dis. Treat. 2018, 14, 567–580. [Google Scholar] [CrossRef] [PubMed]
- McCance-Katz, E.F.; Sullivan, L.E.; Nallani, S. Drug interactions of clinical importance among the opioids, methadone and buprenorphine, and other frequently prescribed medications: A review. Am. J. Addict. 2010, 19, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.H.; Wu, S.; Liu, X.; Yang, X.L.; Xu, J.F.; Guan, Y.T.; Dong, Q.; Zheng, S.L.; Jiang, J.M.; Li, S.X.; et al. Comparison of VerifyNow P2Y12 and thrombelastography for assessing clopidogrel response in stroke patients in China. Neurol. Sci. 2016, 37, 277–282. [Google Scholar] [CrossRef]
- Zhou, S.F.; Wang, L.L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef]
- Kenna, J.; Taskar, K.; Battista, C.; Bourdet, D.; Brouwer, K.; Brouwer, K.; Dai, D.; Funk, C.; Hafey, M.; Lai, Y.; et al. Can bile salt export pump inhibition testing in drug discovery and development reduce liver injury risk? an international transporter consortium perspective. Clin. Pharm. Ther. 2018, 104, 916–932. [Google Scholar] [CrossRef]
- Lepist, E.I.; Ray, A.S. Renal transporter-mediated drug-drug interactions: Are they clinically relevant? J. Clin. Pharmacol. 2016, 56 (Suppl. 7), S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Kearney, B.P.; Flaherty, J.F.; Shah, J. Tenofovir disoproxil fumarate: Clinical pharmacology and pharmacokinetics. Clin. Pharm. 2004, 43, 595–612. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Montoya-Ferrer, A.; Sanz, A.B.; Sanchez-Nino, M.D.; Izquierdo, M.C.; Poveda, J.; Sainz-Prestel, V.; Ortiz-Martin, N.; Parra-Rodriguez, A.; Selgas, R.; et al. Tenofovir nephrotoxicity: 2011 update. AIDS Res. Treat. 2011, 2011, 354908. [Google Scholar] [CrossRef] [PubMed]
- Jackson Rudd, D.; Cao, Y.; Vaddady, P.; Grobler, J.A.; Asante-Appiah, E.; Diamond, T.; Klopfer, S.; Grandhi, A.; Hwang, C.; Vargo, R. Modeling-Supported Islatravir Dose Selection for Phase 3. In Proceedings of the Conference on Retroviruses and Opportunistic Infections (CROI), Boston, MA, USA, 8–11 March 2020. [Google Scholar]
- Matthews, R.; Ankrom, W.; Friedman, E.; Jackson Rudd, D.; Liu, Y.; Mogg, R.; Panebianco, D.; De Lepeleire, I.; Petkova, M.; Grobler, J.; et al. Safety, tolerability, and pharmacokinetics of single- and multiple-dose administration of islatravir (MK-8591) in adults without HIV. Clin. Trans. Sci. 2021, in press. [Google Scholar]
- Matthews, R.; Jackson Rudd, D.; Fillgrove, K.; Zhang, S.; Tomek, C.; Stoch, S.A.; Iwamoto, M. A phase 1 study to evaluate the drug interaction between islatravir (MK-8591) and doravirine in adults without HIV. Clin. Drug Investig. 2021, in press. [Google Scholar] [CrossRef]
- Reese, M.J.; Savina, P.M.; Generaux, G.T.; Tracey, H.; Humphreys, J.E.; Kanaoka, E.; Webster, L.O.; Harmon, K.A.; Clarke, J.D.; Polli, J.W. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 2013, 41, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Ankrom, W.; Jonathan, D.; Jackson Rudd, D.; Zhang, S.; Gravesande, K.; Matthews, R.; Brimhall, D.; Stoch, S.A.; Iwamoto, M. MK-8591 Does Not Alter the Pharmacokinetics of the Oral Contraceptives Levonorgestrel and Ethinyl Estradiol. In Proceedings of the IDWeek 2018, San Francisco, CA, USA, 3–7 October 2018. [Google Scholar]
- Pfizer Inc. ALESSE® [Product Information]; Pfizer Canada Inc.: Kirkland, QC, Canada, 2017. [Google Scholar]
- US Food and Drug Administration Center for Drug Evaluation and Research. In Vitro Metabolism- and Transporter-Mediated Drug-Drug Interaction Studies Guidance for Industry. 2017. Available online: https://www.fda.gov/media/108130/download (accessed on 22 July 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Renal Clearance (mL/min/kg) | Renal Excretion (% Total Plasma Clearance) |
|---|---|---|
| Mouse | 15.4 | 61 |
| Rat | 11.5 | 17 |
| Rabbit | 5.3 | 31 |
| Rhesus macaque | 8.7 | 51 |
| Islatravir Risk of Interaction with Metabolic Enzymes | |||||
| Enzyme | Mechanism of Inhibition | Islatravir IC50 (µM) a | Maximum Unbound Plasma Concentration b (Imax,u) to Ki,u c Ratio (µM) | Intestinal Concentration d (Igut) to Ki,u c Ratio (µM) | DDI Potential f |
| CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 | Reversible | >100 | <0.019 | N/A | Low risk |
| CYP3A4 | Reversible | >200 | <0.010 | <8.2 | Low risk |
| UGT1A1 | Reversible | >100 | N/A | <16.4 | Low risk g |
| CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4 | Time dependent | >50 | N/A | N/A | Low risk h |
| Islatravir Risk of Interaction with Drug Transporters | |||||
| Transporter | Islatravir IC50 (µM) a | Maximum Unbound Plasma Concentration b (Imax,u) to IC50 Ratio (µM) | Intestinal Concentration d (Igut) to IC50 Ratio (µM) | Maximum Unbound Inlet Concentration e (Iin,max,u) to IC50 Ratio | DDI Potential f |
| OATP1B1, OATP1B3, OCT1 | >300 | N/A | N/A | <0.035 | Low risk |
| OAT1, OAT3, OCT2 | >100 | <0.010 | N/A | N/A | Low risk |
| MATE1, MATE2K | >75 | <0.013 | N/A | N/A | Low risk |
| BCRP | >100 | <0.010 | <8.2 | N/A | Low risk |
| MDR1 P-gp | >200 | <0.005 | <4.1 | N/A | Low risk |
| Concentration [µM] | mRNA Mean Fold Change ± SD a | |||
|---|---|---|---|---|
| CYP3A4 | CYP2B6 | CYP1A2 | ||
| DMSO (vehicle) | NA | 1.0 ± 0.0 | 1.0 ± 0.0 | 1.0 ± 0.0 |
| Rifampin (control) | 10 | 9.9 ± 2.7 | ND | ND |
| Phenobarbitol (control) | 1000 | ND | 18.5 ± 1.9 | ND |
| Omeprazole (control) | 50 | ND | ND | 26.4 ± 8.6 |
| Islatravir | 0.1 | 0.6 ± 0.2 | 0.5 ± 0.1 | 0.4 ± 0.2 |
| 0.5 | 0.6 ± 0.2 | 0.5 ± 0.2 | 0.4 ± 0.2 | |
| 1 | 0.6 ± 0.2 | 0.7 ± 0.2 | 0.5 ± 0.3 | |
| 5 | 0.5 ± 0.1 | 0.7 ± 0.1 | 0.4 ± 0.3 | |
| 10 | 0.6 ± 0.1 | 0.9 ± 0.3 | 0.5 ± 0.4 | |
| 20 | 0.1 ± 0.1 | 0.4 ± 0.3 | 0.2 ± 0.2 | |
| Transporter | Islatravir Uptake a (pmole/106 Cells) | Fold-Difference b | Conclusions | |
|---|---|---|---|---|
| Control Cells | Transporter-Expressing Cells | |||
| OCT2 | 0.97 ± 0.01 | 0.79 ± 0.14 | 0.81 | Non-substrate |
| OAT1 | 0.69 ± 0.07 | 0.72 ± 0.04 | 1.04 | Non-substrate |
| OAT3 | 0.69 ± 0.07 | 0.85 ± 0.06 | 1.23 | Non-substrate |
| MATE1 | 2.90 ± 0.27 | 2.94 ± 0.20 | 1.01 | Non-substrate |
| MATE2K | 3.12 ± 0.17 | 3.56 ± 0.17 | 1.14 | Non-substrate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bleasby, K.; Houle, R.; Hafey, M.; Lin, M.; Guo, J.; Lu, B.; Sanchez, R.I.; Fillgrove, K.L. Islatravir Is Not Expected to Be a Victim or Perpetrator of Drug-Drug Interactions via Major Drug-Metabolizing Enzymes or Transporters. Viruses 2021, 13, 1566. https://doi.org/10.3390/v13081566
Bleasby K, Houle R, Hafey M, Lin M, Guo J, Lu B, Sanchez RI, Fillgrove KL. Islatravir Is Not Expected to Be a Victim or Perpetrator of Drug-Drug Interactions via Major Drug-Metabolizing Enzymes or Transporters. Viruses. 2021; 13(8):1566. https://doi.org/10.3390/v13081566
Chicago/Turabian StyleBleasby, Kelly, Robert Houle, Michael Hafey, Meihong Lin, Jingjing Guo, Bing Lu, Rosa I. Sanchez, and Kerry L. Fillgrove. 2021. "Islatravir Is Not Expected to Be a Victim or Perpetrator of Drug-Drug Interactions via Major Drug-Metabolizing Enzymes or Transporters" Viruses 13, no. 8: 1566. https://doi.org/10.3390/v13081566
APA StyleBleasby, K., Houle, R., Hafey, M., Lin, M., Guo, J., Lu, B., Sanchez, R. I., & Fillgrove, K. L. (2021). Islatravir Is Not Expected to Be a Victim or Perpetrator of Drug-Drug Interactions via Major Drug-Metabolizing Enzymes or Transporters. Viruses, 13(8), 1566. https://doi.org/10.3390/v13081566