
The IGF-1 Signaling Pathway in Viral Infections



{kind=link}
Abstract
:1. Introduction
2. IGF-1 and IGF-1R
3. IGFBPs
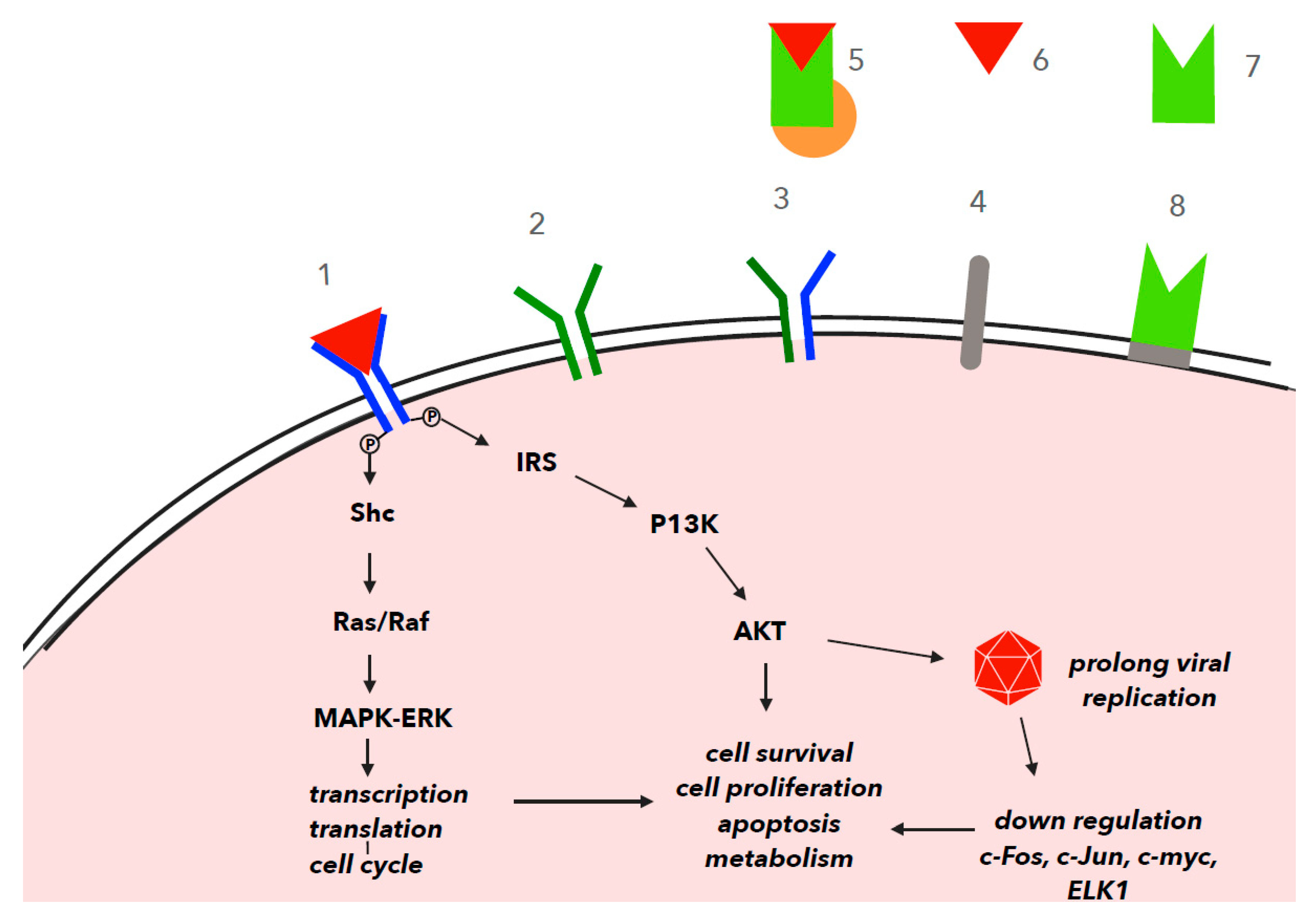
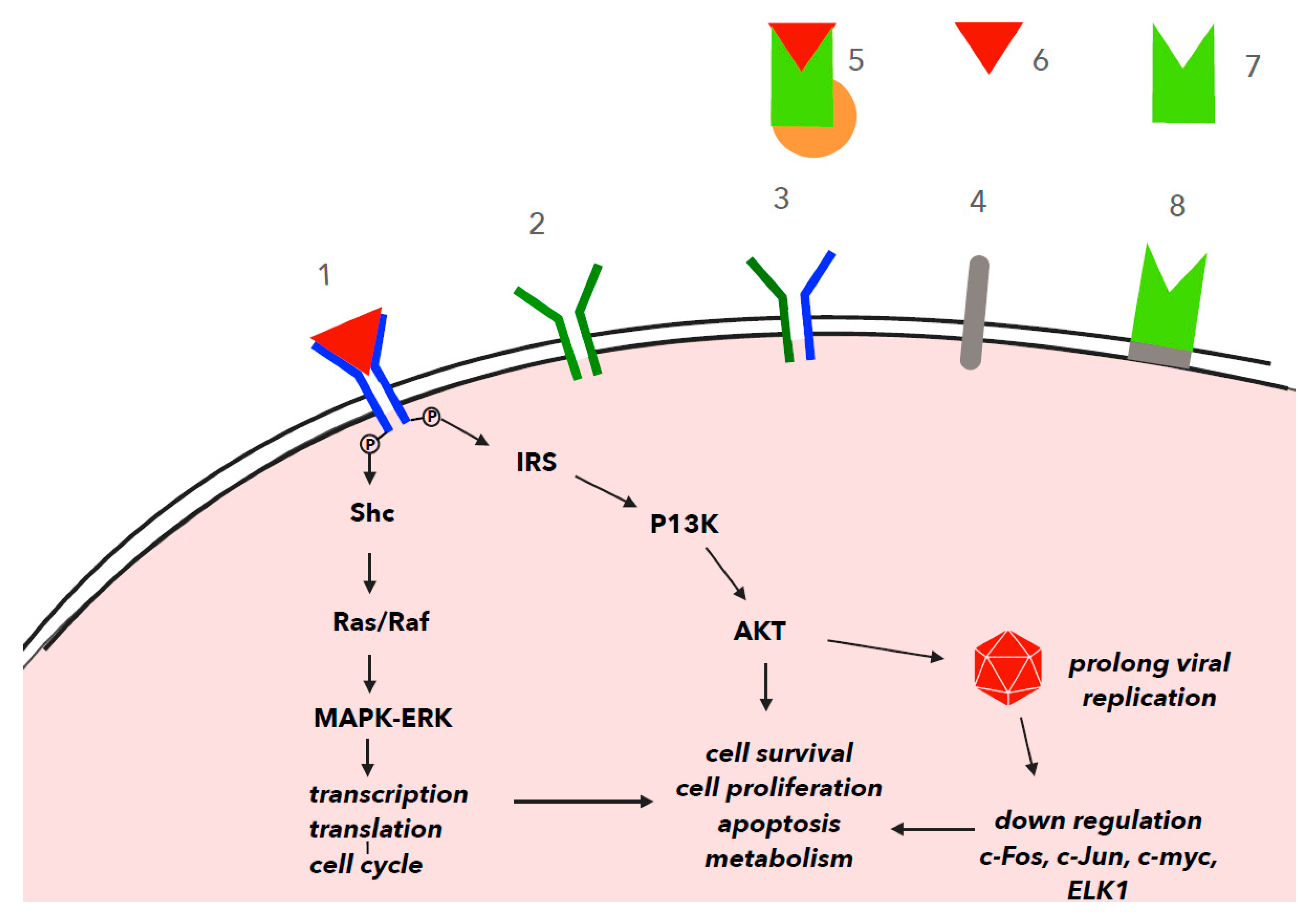
4. IGF-1 in Cell Signaling and Viral Infection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Gusscott, S.; Tamiro, F.; Giambra, V.; Weng, A.P. Insulin-like growth factor (IGF) signaling in T-cell acute lymphoblastic leukemia. Adv. Biol. Regul. 2019, 74, 100652. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses 2020, 12, 977. [Google Scholar] [CrossRef]
- Ji, W.-T.; Liu, H. PI3K-Akt Signaling and Viral Infection. Recent Pat. Biotechnol. 2008, 2, 218–226. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- DuShane, J.K.; Wilczek, M.P.; Mayberry, C.L.; Maginnis, M.S. ERK Is a Critical Regulator of JC Polyomavirus Infection. J. Virol. 2018, 92, e01529-17. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Hou, X.; Peng, H.; Zhang, L.; Li, Y.; Gu, Z.; Jiang, Q.; Shi, M.; Ji, Y.; Jiang, J. MEK/ERK signaling pathway is required for enterovirus 71 replication in immature dendritic cells. Virol. J. 2014, 11, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczorowska, M.M.; Kwasniewska, A.; Gozdzicka-Jozefiak, A. IGF1 mRNA isoform expression in the cervix of HPV-positive women with pre-cancerous and cancer lesions. Exp. Ther. Med. 2011, 2, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Merle, P.; Trepo, C. Molecular Mechanisms Underlying Hepatocellular Carcinoma. Viruses 2009, 1, 852–872. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, R.J.; LeBeau, A.P.; Fulmer, C.G.; Lazzarino, D.A.; Hochberg, A.; Wood, T.L. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J. Biol. Chem. 2007, 282, 22513–22524. [Google Scholar] [CrossRef] [Green Version]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocmé, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef]
- Solomon-Zemler, R.; Sarfstein, R.; Werner, H. Nuclear insulin-like growth factor-1 receptor (IGF1R) displays proliferative and regulatory activities in non-malignant cells. PLoS ONE 2017, 12, e0185164. [Google Scholar] [CrossRef] [Green Version]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warsito, D.; Sjöström, S.; Andersson, S.; Larsson, O.; Sehat, B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep. 2012, 13, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Arainga, M.; Takeda, E.; Aida, Y. Identification of bovine leukemia virus tax function associated with host cell transcription, signaling, stress response and immune response pathway by microarray-based gene expression analysis. BMC Genom. 2012, 13, 121. [Google Scholar] [CrossRef] [Green Version]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef]
- Dale, O.T.; Aleksic, T.; Shah, K.A.; Han, C.; Mehanna, H.; Rapozo, D.C.M.; Sheard, J.D.H.; Goodyear, P.; Upile, N.S.; Robinson, M.; et al. IGF-1R expression is associated with HPV-negative status and adverse survival in head and neck squamous cell cancer. Carcinogenesis 2015, 36, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Yahya, M.A.; Sharon, S.M.; Hantisteanu, S.; Hallak, M.; Bruchim, I. The role of the insulin-like growth factor 1 pathway in immune tumor microenvironment and its clinical ramifications in gynecologic malignancies. Front. Endocrinol. 2018, 9, 297. [Google Scholar] [CrossRef]
- Nahor, I.; Abramovitch, S.; Engeland, K.; Werner, H. The p53-family members p63 and p73 inhibit insulin-like growth factor-I receptor gene expression in colon cancer cells. Growth Horm. IGF Res. 2005, 15, 388–396. [Google Scholar] [CrossRef]
- Abramovitch, S.; Glaser, T.; Ouchi, T.; Werner, H. BRCA1-Sp1 interactions in transcriptional regulation of the IGF-IR gene. FEBS Lett. 2003, 541, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Jozefiak, A.; Pacholska-Bogalska, J.; Myga-Nowak, M.; Kedzia, W.; Kwasniewska, A.; Luczak, M.; Kedzia, H.; Gozdzicka-Jozefiak, A. Serum and tissue levels of insulin-like growth factor-I in women with dysplasia and HPV-positive cervical cancer. Mol. Med. Rep. 2008, 1, 231–237. [Google Scholar]
- Li, C.; Harada, A.; Oh, Y. IGFBP-3 sensitizes antiestrogen-resistant breast cancer cells through interaction with GRP78. Cancer Lett. 2012, 325, 200–206. [Google Scholar] [CrossRef]
- Lodhia, K.A.; Tienchaiananda, P.; Haluska, P. Understanding the Key to Targeting the IGF Axis in Cancer: A Biomarker Assessment. Front. Oncol. 2015, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Lee, H.Y.; Weinzimer, S.A.; Powell, D.R.; Clifford, J.L.; Kurie, J.M.; Cohen, P. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-α regulate transcriptional signaling and apoptosis. J. Biol. Chem. 2000, 275, 33607–33613. [Google Scholar] [CrossRef] [Green Version]
- Grkovic, S.; O’Reilly, V.C.; Han, S.; Hong, M.; Baxter, R.C.; Firth, S.M. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene 2013, 32, 2412–2420. [Google Scholar] [CrossRef] [Green Version]
- Matilainen, M.; Malinen, M.; Saavalainen, K.; Carlberg, C. Regulation of multiple insulin-like growth factor binding protein genes by 1α,25-dihydroxyvitamin D3. Nucleic Acids Res. 2005, 33, 5521–5532. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Ide, S.; Takashima, S.; Kudo, S.; Nomura, Y.; Segawa, M.; Kubota, T.; Mori, H.; Tanaka, S.; Horie, H.; et al. Methyl CpG-Binding Protein 2 (a Mutation of Which Causes Rett Syndrome) Directly Regulates Insulin-Like Growth Factor Binding Protein 3 in Mouse and Human Brains. J. Neuropathol. Exp. Neurol. 2007, 66, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Azar, W.J.; Zivkovic, S.; Werther, G.A.; Russo, V.C. IGFBP-2 nuclear translocation is mediated by a functional NLS sequence and is essential for its pro-tumorigenic actions in cancer cells. Oncogene 2014, 33, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.K.; Hu, L.; Fuller, G.N.; Zhang, W. An interaction between insulin-like growth factor-binding protein 2 (IGFBP2) and integrin α5 is essential for IGFBP2-induced cell mobility. J. Biol. Chem. 2006, 281, 14085–14091. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; Yang, Z.; Bach, L.A. Prohibitin-2 binding modulates insulin-like growth factor-binding protein-6 (IGFBP-6)-induced rhabdomyosarcoma cell migration. J. Biol. Chem. 2013, 288, 29890–29900. [Google Scholar] [CrossRef] [Green Version]
- Weigel, K.J.; Jakimenko, A.; Conti, B.A.; Chapman, S.E.; Kaliney, W.J.; Leevy, W.M.; Champion, M.M.; Schafer, Z.T. CAF-Secreted IGFBPs Regulate Breast Cancer Cell Anoikis. Mol. Cancer Res. 2014, 12, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Diehl, N.; Schaal, H. Make yourself at home: Viral hijacking of the PI3K/Akt signaling pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, C.D.; Bilawchuk, L.M.; McDonough, J.E.; Jamieson, K.C.; Elawar, F.; Cen, Y.; Duan, W.; Lin, C.; Song, H.; Casanova, J.-L.; et al. IGF1R is an entry receptor for respiratory syncytial virus. Nature 2020, 583, 615–619. [Google Scholar] [CrossRef]
- Kumar, D.; Broor, S.; Rajala, M.S. Interaction of Host Nucleolin with Influenza A Virus Nucleoprotein in the Early Phase of Infection Limits the Late Viral Gene Expression. PLoS ONE 2016, 11, e0164146. [Google Scholar] [CrossRef]
- Honda, T.; Fujino, K.; Okuzaki, D.; Ohtaki, N.; Matsumoto, Y.; Horie, M.; Daito, T.; Itoh, M.; Tomonaga, K. Upregulation of insulin-like growth factor binding protein 3 in astrocytes of transgenic mice that express Borna disease virus phosphoprotein. J. Virol. 2011, 85, 4567–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elawar, F.; Griffiths, C.D.; Zhu, D.; Bilawchuk, L.M.; Jensen, L.D.; Forss, L.; Tang, J.; Hazes, B.; Drews, S.J.; Marchant, D.J. A Virological and Phylogenetic Analysis of the Emergence of New Clades of Respiratory Syncytial Virus. Sci. Rep. 2017, 7, 12232. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, D.; Eizuru, Y.; Tokunaga, M.; Takada, K. Autocrine Growth of Epstein-Barr Virus-Positive Gastric Carcinoma Cells Mediated by an Epstein-Barr Virus-Encoded Small RNA. Cancer Res. 2003, 63, 7062–7067. [Google Scholar]
- Winn, B.J. Is there a role for insulin-like growth factor inhibition in the treatment of COVID-19-related adult respiratory distress syndrome? Med. Hypotheses 2020, 144, 110167. [Google Scholar] [CrossRef]
- Altindis, E.; Cai, W.; Sakaguchi, M.; Zhang, F.; GuoXiao, W.; Liu, F.; De Meyts, P.; Gelfanov, V.; Pan, H.; DiMarchi, R.; et al. Viral insulin-like peptides activate human insulin and IGF-1 receptor signaling: A paradigm shift for host-microbe interactions. Proc. Natl. Acad. Sci. USA 2018, 115, 2461–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carotti, S.; Guarino, M.P.L.; Valentini, F.; Porzio, S.; Vespasiani-Gentilucci, U.; Perrone, G.; Zingariello, M.; Gallo, P.; Cicala, M.; Picardi, A.; et al. Impairment of GH/IGF-1 Axis in the Liver of Patients with HCV-Related Chronic Hepatitis. Horm. Metab. Res. 2018, 50, 145–151. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.-C.; Deng, M.; Jiang, H.-Y.; Guo, L.-H.; Zhou, W.-J.; Ruan, B. Serum insulin-like growth factor-1 and its binding protein 3 as prognostic factors for the incidence, progression, and outcome of hepatocellular carcinoma: A systematic review and meta-analysis. Oncotarget 2017, 8, 81098–81108. [Google Scholar] [CrossRef] [Green Version]
- Scharf, J.G.; Ramadori, G.; Dombrowski, F. Analysis of the IGF axis in preneoplastic hepatic foci and hepatocellular neoplasms developing after low-number pancreatic islet transplantation into the livers of streptozotocin diabetic rats. Lab. Investig. 2000, 80, 1399–1411. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J. Multisite protein modification and intramolecular signaling. Oncogene 2005, 24, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Crow, M.S.; Lum, K.K.; Sheng, X.; Song, B.; Cristea, I.M. Diverse mechanisms evolved by DNA viruses to inhibit early host defenses. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 452–481. [Google Scholar] [CrossRef] [PubMed]
- Wojcechowskyj, J.A.; Didigu, C.A.; Lee, J.Y.; Parrish, N.F.; Sinha, R.; Hahn, B.H.; Bushman, F.D.; Jensen, S.T.; Seeholzer, S.H.; Doms, R.W. Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe 2013, 13, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubiec, A.; Jupin, I. Regulation of positive-strand RNA virus replication: The emerging role of phosphorylation. Virus Res. 2007, 129, 73–79. [Google Scholar] [CrossRef]
- Keating, J.A.; Striker, R. Phosphorylation events during viral infections provide potential therapeutic targets. Rev. Med. Virol. 2012, 22, 166–181. [Google Scholar] [CrossRef]
- Yu, Y.; Alwine, J.C. Human Cytomegalovirus Major Immediate-Early Proteins and Simian Virus 40 Large T Antigen Can Inhibit Apoptosis through Activation of the Phosphatidylinositide 3′-OH Kinase Pathway and the Cellular Kinase Akt. J. Virol. 2002, 76, 3731–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.-N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 2015, 14, 87. [Google Scholar] [CrossRef] [Green Version]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accardi, R.; Rubino, R.; Scalise, M.; Gheit, T.; Shahzad, N.; Thomas, M.; Banks, L.; Indiveri, C.; Sylla, B.S.; Cardone, R.A.; et al. E6 and E7 from Human Papillomavirus Type 16 Cooperate To Target the PDZ Protein Na/H Exchange Regulatory Factor 1. J. Virol. 2011, 85, 8208–8216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.H.; Wu, J.Y.; Cheng, Y.W.; Chen, C.Y.; Lee, M.C.; Goan, Y.G.; Lee, H. cIAP2 upregulated by E6 oncoprotein via epidermal growth factor receptor/phosphatidylinositol 3-kinase/AKT pathway confers resistance to cisplatin in human papillomavirus 16/18-infected lung cancer. Clin. Cancer Res. 2010, 16, 5200–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chen, J.; Zhang, L.; Zhao, P.P.M.; Zhao, K.-N. Four Major Factors Regulate Phosphatidylinositol 3-kinase Signaling Pathway in Cancers Induced by Infection of Human Papillomaviruses. Curr. Med. Chem. 2014, 21, 3057–3069. [Google Scholar] [CrossRef]
- Charette, S.T.; McCance, D.J. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an Akt-dependent manner. Oncogene 2007, 26, 7386. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Liefers-Visser, J.A.L.; Meijering, R.A.M.; Reyners, A.K.L.; van der Zee, A.G.J.; de Jong, S. IGF system targeted therapy: Therapeutic opportunities for ovarian cancer. Cancer Treat. Rev. 2017, 60, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachos, G.; Koffa, M.; Preston, C.M.; Clements, J.B.; Conner, J. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J. Virol. 2001, 75, 2710–2728. [Google Scholar] [CrossRef] [Green Version]
- Pétigny-Lechartier, C.; Duboc, C.; Jebahi, A.; Louis, M.-H.; Abeilard, E.; Denoyelle, C.; Gauduchon, P.; Poulain, L.; Villedieu, M. The mTORC1/2 Inhibitor AZD8055 Strengthens the Efficiency of the MEK Inhibitor Trametinib to Reduce the Mcl-1/[Bim and Puma] ratio and to Sensitize Ovarian Carcinoma Cells to ABT-737. Mol. Cancer Ther. 2017, 16, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, L.A.; Kruse, E.A.; Puthalakath, H.; Kelly, P.N.; Kaufmann, T.; Huang, D.C.S.; Strasser, A. MEK/ERK-mediated phosphorylation of Bim is required to ensure survival of T and B lymphocytes during mitogenic stimulation. J. Immunol. 2009, 183, 261–269. [Google Scholar] [CrossRef]
- Moustafa-Kamal, M.; Gamache, I.; Lu, Y.; Li, S.; Teodoro, J.G. BimEL is phosphorylated at mitosis by Aurora A and targeted for degradation by βTrCP1. Cell Death Differ. 2013, 20, 1393–1403. [Google Scholar] [CrossRef]
- Deng, J. How to unleash mitochondrial apoptotic blockades to kill cancers? Acta Pharm. Sin. B 2017, 7, 18–26. [Google Scholar] [CrossRef]
- Monick, M.M.; Cameron, K.; Staber, J.; Powers, L.S.; Yarovinsky, T.O.; Koland, J.G.; Hunninghake, G.W. Activation of the Epidermal Growth Factor Receptor by Respiratory Syncytial Virus Results in Increased Inflammation and Delayed Apoptosis. J. Biol. Chem. 2005, 280, 2147–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Kadota, S.; Takeda, M.; Miyajima, N.; Nagata, K. Measles virus V protein blocks interferon (IFN)-α/β but not IFN-γ signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003, 545, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Bordignon, V.; Di Domenico, E.; Trento, E.; D’Agosto, G.; Cavallo, I.; Pontone, M.; Pimpinelli, F.; Mariani, L.; Ensoli, F. How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses 2017, 9, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Józefiak, A.; Larska, M.; Pomorska-Mól, M.; Ruszkowski, J.J. The IGF-1 Signaling Pathway in Viral Infections. Viruses 2021, 13, 1488. https://doi.org/10.3390/v13081488
Józefiak A, Larska M, Pomorska-Mól M, Ruszkowski JJ. The IGF-1 Signaling Pathway in Viral Infections. Viruses. 2021; 13(8):1488. https://doi.org/10.3390/v13081488
Chicago/Turabian StyleJózefiak, Agata, Magdalena Larska, Małgorzata Pomorska-Mól, and Jakub J. Ruszkowski. 2021. "The IGF-1 Signaling Pathway in Viral Infections" Viruses 13, no. 8: 1488. https://doi.org/10.3390/v13081488
APA StyleJózefiak, A., Larska, M., Pomorska-Mól, M., & Ruszkowski, J. J. (2021). The IGF-1 Signaling Pathway in Viral Infections. Viruses, 13(8), 1488. https://doi.org/10.3390/v13081488