
Rotavirus A Genome Segments Show Distinct Segregation and Codon Usage Patterns


Abstract
1. Introduction
1.1. Rotavirus A Genome and Proteins
1.2. Selective Pressures on Synonymous Sites
2. Materials and Methods
2.1. Sequence Alignment and Phylogenetic Analysis
2.2. Codon Bias Analysis Comparison
3. Results
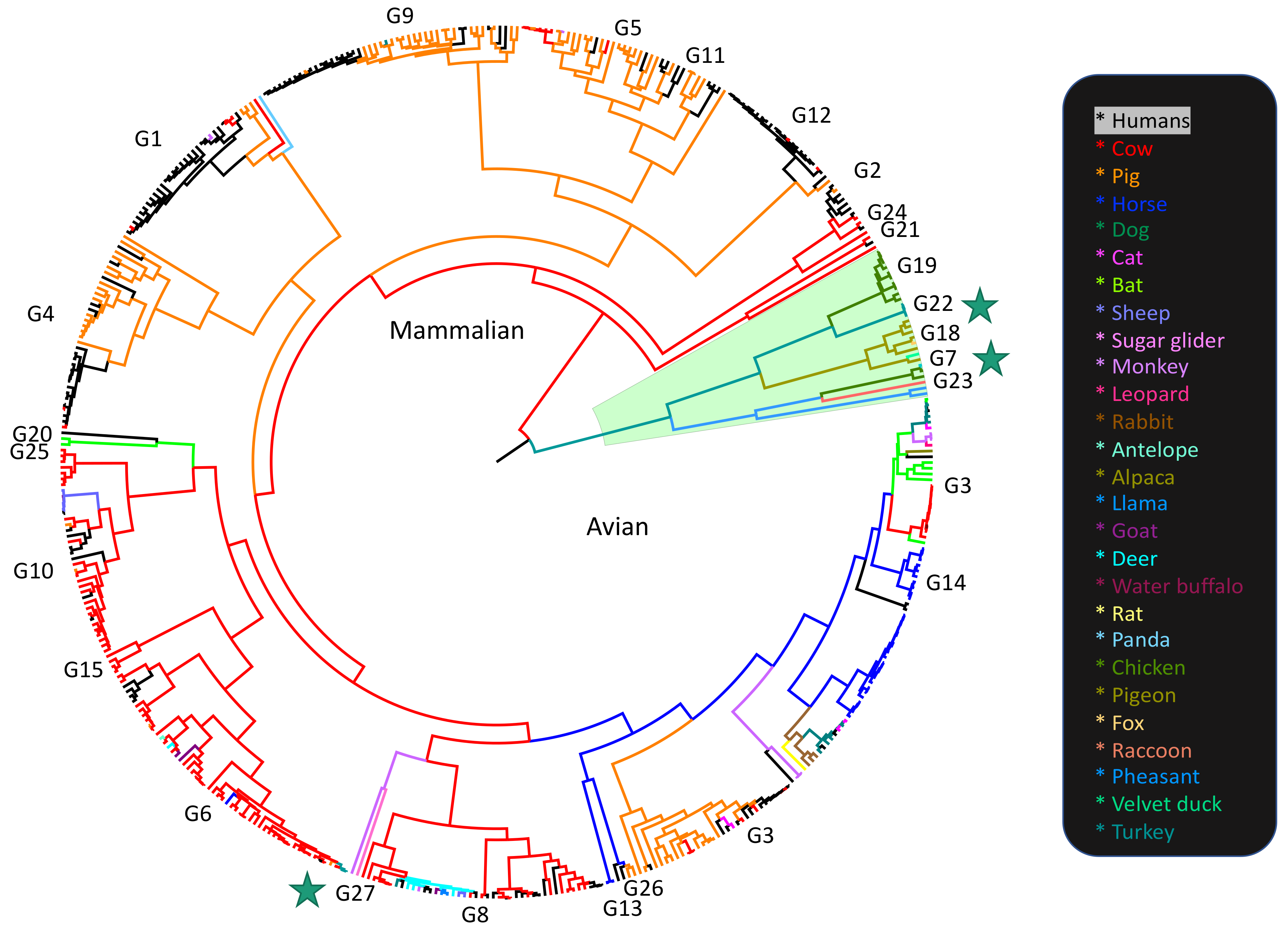
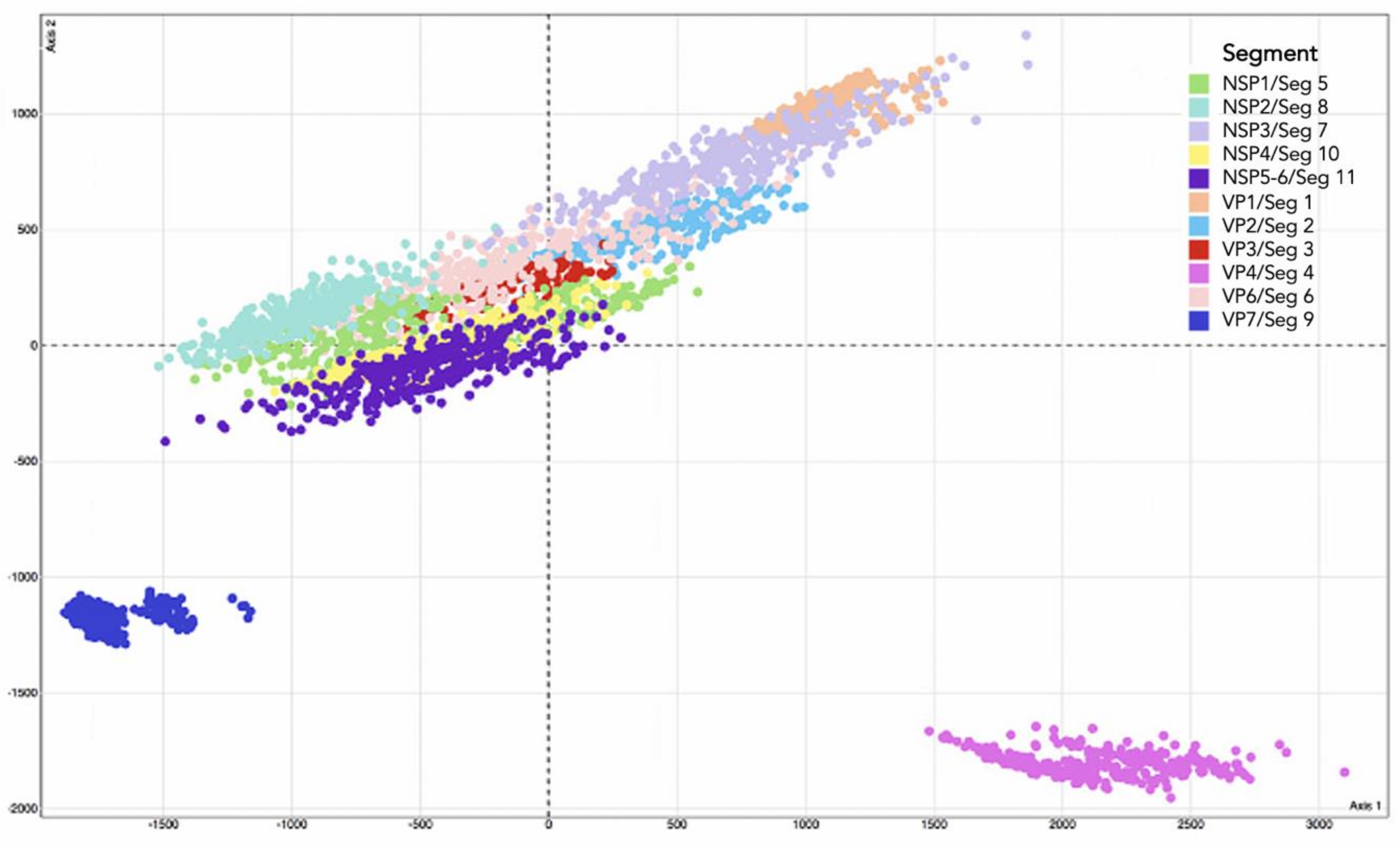
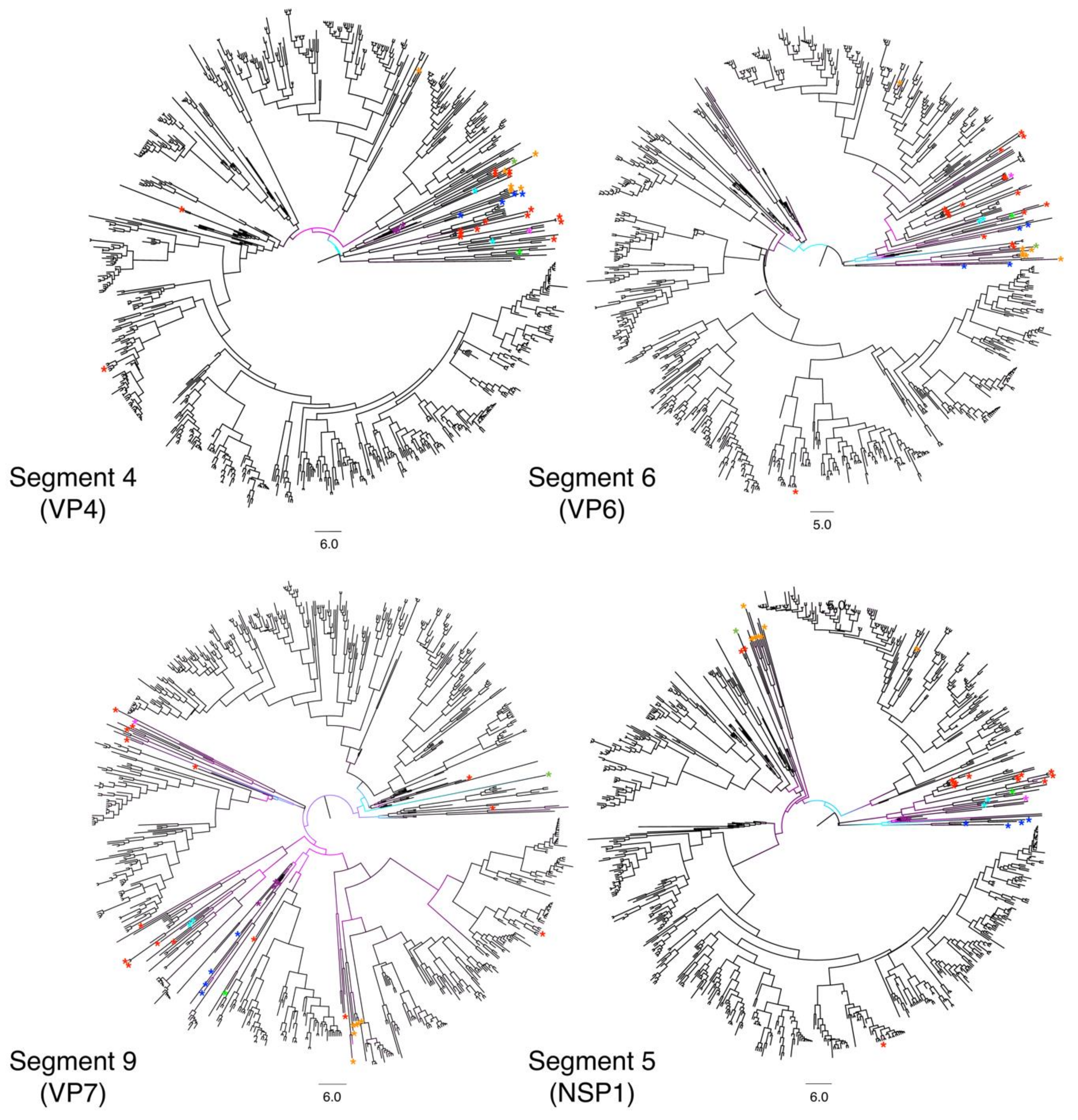
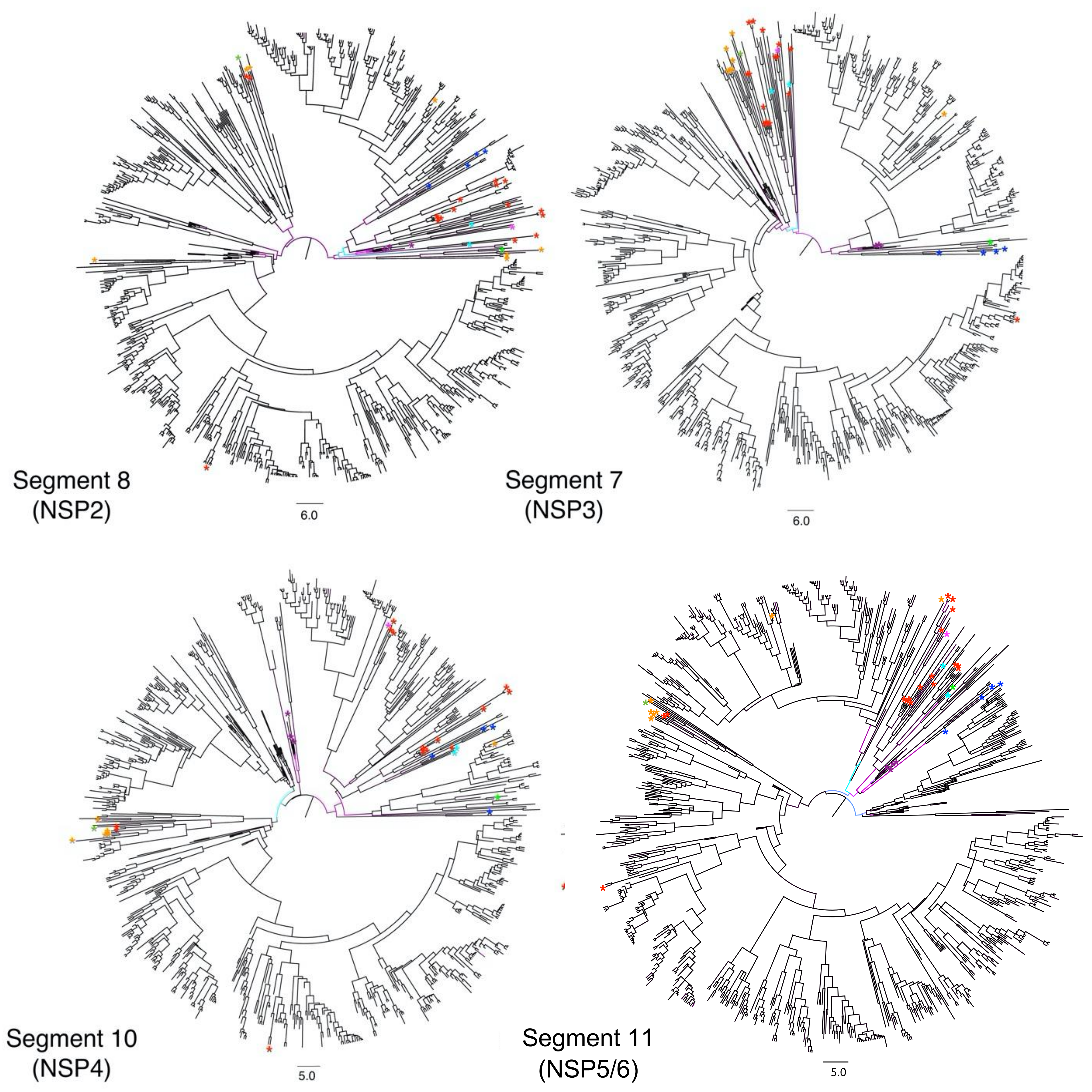
3.1. Different Tree Space Occupied by the 11 Segments
3.2. Evolutionary Rates
3.3. Differing Codon Usage Patterns by Segment
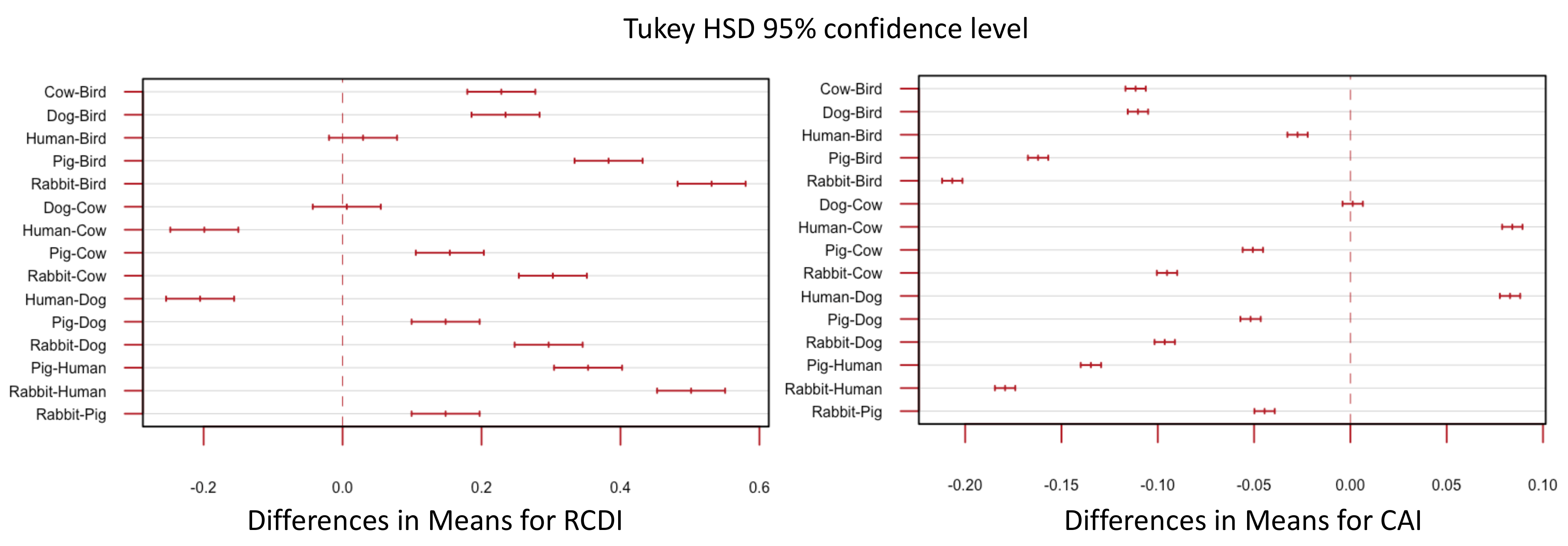
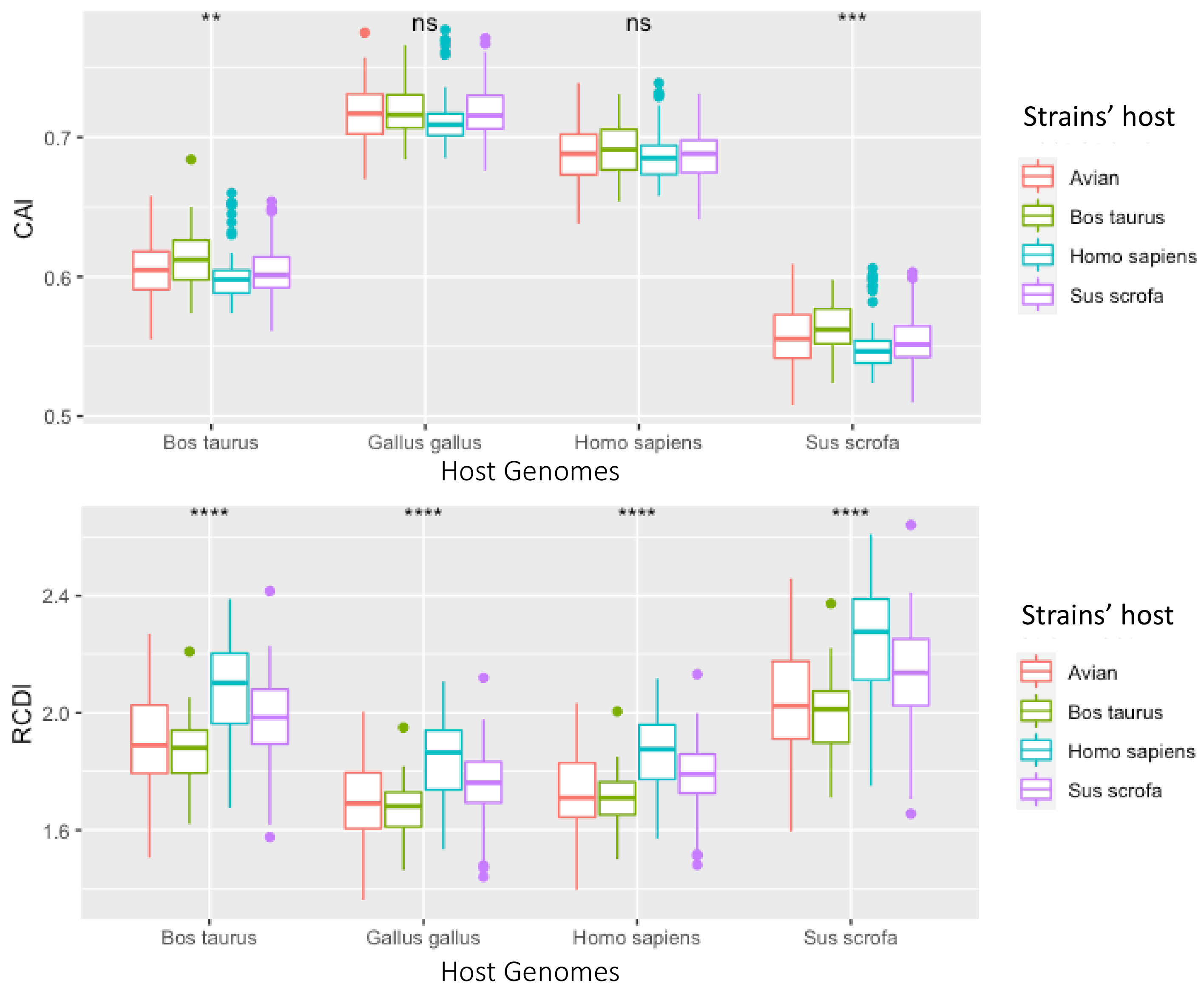
3.4. Synonymous Sites under Selection, However Evidence Does Not Suggest Translational Selection to Specific Hosts
4. Discussion
4.1. Potential Impact of Rotavirus RNA and Protein Interactions on Segment Co-Segregation
4.2. Rotavirus Evolution Following the Introduction of Rotavirus Vaccines
4.3. Codon Bias Analysis Shows Codon Usage Differs between Segments, but Not between Host Strains
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Bilcke, J.; Ciarlet, M.; Martella, V.; Bányai, K.; Rahman, M.; Zeller, M.; Beutels, P.; Van Damme, P.; Van Ranst, M. Rotavirus disease and vaccination: Impact on genotype diversity. Future Microbiol. 2009, 4, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.; et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Banyai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gomara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Trojnar, E.; Heckel, G.; Otto, P.H.; Johne, R. Analysis of rotavirus species diversity and evolution including the newly determined full-length genome sequences of rotavirus F and G. Infect. Genet. Evol. 2013, 14, 58–67. [Google Scholar] [CrossRef]
- Johne, R.; Reetz, J.; Kaufer, B.B.; Trojnar, E. Generation of an Avian-Mammalian Rotavirus Reassortant by Using a Helper Virus-Dependent Reverse Genetics System. J. Virol. 2016, 90, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.M.; Gregori, F.; Souza, S.P.; Rotava, D.; Oliveira, R.N.; Villarreal, L.Y.B.; Richtzenhain, L.J.; Brandão, P.E. Bovine rotavirus in turkeys with enteritis. Avian Dis. 2011, 55, 697–700. [Google Scholar] [CrossRef]
- Brüssow, H.; Nakagomi, O.; Gerna, G.; Eichhorn, W. Isolation of an avianlike group A rotavirus from a calf with diarrhea. J. Clin. Microbiol. 1992, 30, 67–73. [Google Scholar] [CrossRef]
- Busi, C.; Martella, V.; Papetti, A.; Sabelli, C.; Lelli, D.; Alborali, G.L.; Gibelli, L.; Gelmetti, D.; Lavazza, A.; Cordioli, P.; et al. Group A Rotavirus Associated with Encephalitis in Red Fox. Emerg. Infect. Dis. 2017, 23, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Trojnar, E.; Sachsenroder, J.; Twardziok, S.; Reetz, J.; Otto, P.H.; Johne, R. Identification of an avian group A rotavirus con-taining a novel VP4 gene with a close relationship to those of mammalian rotaviruses. J. Gen. Virol. 2013, 94, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kool, D.A.; Matsui, S.M.; Greenberg, H.B.; Holmes, I.H. Isolation and characterization of a novel reassortant between avian Ty-1 and simian RRV rotaviruses. J. Virol. 1992, 66, 6836–6839. [Google Scholar] [CrossRef] [PubMed]
- Patzina-Mehling, C.; Falkenhagen, A.; Trojnar, E.; Gadicherla, A.K.; Johne, R. Potential of avian and mammalian species A rotaviruses to reassort as explored by plasmid only-based reverse genetics. Virus Res. 2020, 286, 198027. [Google Scholar] [CrossRef] [PubMed]
- Schoondermark-van de Ven, E.; Van Ranst, M.; de Bruin, W.; van den Hurk, P.; Zeller, M.; Matthijnssens, J.; Heylen, E. Rabbit colony infected with a bovine-like G6P[11] rotavirus strain. Vet. Microbiol. 2013, 166, 154–164. [Google Scholar] [CrossRef]
- .Heylen, E.; Likele, B.B.; Zeller, M.; Stevens, S.; De Coster, S.; Conceição-Neto, N.; Van Geet, C.; Jacobs, J.; Ngbonda, D.; Van Ranst, M.; et al. Rotavirus surveillance in Kisangani, the Democratic Republic of the Congo, reveals a high number of unusual genotypes and gene segments of animal origin in non-vaccinated symptomatic children. PLoS ONE 2014, 9, e100953. [Google Scholar] [CrossRef]
- Xia, L.; Fan, Q.; He, B.; Xu, L.; Zhang, F.; Hu, T.; Wang, Y.; Li, N.; Qiu, W.; Zheng, Y.; et al. The complete genome sequence of a G3P[10] Chinese bat rotavirus suggests multiple bat rotavirus inter-host species transmission events. Infect. Genet. Evol. 2014, 28, 1–4. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Rahman, M.; Martella, V.; Xuelei, Y.; De Vos, S.; De Leener, K.; Ciarlet, M.; Buonavoglia, C.; Van Ranst, M. Full genomic analysis of human rotavirus strain B4106 and lapine rotavirus strain 30/96 provides evidence for interspecies transmission. J. Virol. 2006, 80, 3801–3810. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Taraporewala, Z.F.; Yang, H.; Rao, S.; Yuan, L.; Cao, D.; Hoshino, Y.; Mertens, P.; Carner, G.R.; McNeal, M.; et al. Simian rotaviruses possess divergent gene constellations that originated from interspecies transmission and reassortment. J. Virol. 2010, 84, 2013–2026. [Google Scholar] [CrossRef]
- Matthijnssens, J.; De Grazia, S.; Piessens, J.; Heylen, E.; Zeller, M.; Giammanco, G.M.; Bányai, K.; Buonavoglia, C.; Ciarlet, M.; Martella, V.; et al. Multiple reassortment and interspecies transmission events contribute to the diversity of feline, canine and feline/canine-like human group A rotavirus strains. Infect. Genet. Evol. 2011, 11, 1396–1406. [Google Scholar] [CrossRef]
- Bányai, K.; Esona, M.D.; Kerin, T.K.; Hull, J.J.; Mijatovic, S.; Vásconez, N.; Torres, C.; De Filippis, A.M.B.; Gentsch, J.R. Molecular characterization of a rare, human-porcine reassortant rotavirus strain, G11P[6], from Ecuador. Arch. Virol. 2009, 154, 1823–1829. [Google Scholar] [CrossRef]
- Miño, S.; Matthijnssens, J.; Badaracco, A.; Garaicoechea, L.; Zeller, M.; Heylen, E.; Van Ranst, M.; Barrandeguy, M.; Parreño, V. Equine G3P[3] rotavirus strain E3198 related to simian RRV and feline/canine-like rotaviruses based on complete genome analyses. Vet. Microbiol. 2013, 161, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Mullick, S.; Mukherjee, A.; Ghosh, S.; Pazhani, G.P.; Sur, D.; Manna, B.; Nataro, J.P.; Levine, M.M.; Ramamurthy, T.; Chawla-Sarkar, M. Genomic analysis of human rotavirus strains G6P[14] and G11P[25] isolated from Kolkata in 2009 reveals interspecies transmission and complex reassortment events. Infect. Genet. Evol. 2013, 14, 15–21. [Google Scholar] [CrossRef]
- Banyai, K.; Martella, V.; Molnar, P.; Mihaly, I.; Van Ranst, M.; Matthijnssens, J. Genetic heterogeneity in human G6P[14] rotavirus strains detected in Hungary suggests independent zoonotic origin. J. Infect. 2009, 59, 213–215. [Google Scholar] [CrossRef]
- Papp, H.; Borzák, R.; Farkas, S.; Kisfali, P.; Lengyel, G.; Molnár, P.; Melegh, B.; Matthijnssens, J.; Jakab, F.; Martella, V.; et al. Zoonotic transmission of reassortant porcine G4P[6] rotaviruses in Hungarian pediatric patients identified sporadically over a 15 year period. Infect. Genet. Evol. 2013, 19, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Papp, H.; Matthijnssens, J.; Martella, V.; Ciarlet, M.; Bányai, K. Global distribution of group A rotavirus strains in horses: A systematic review. Vaccine 2013, 31, 5627–5633. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-I.; Matthijnssens, J.; Saif, L.J.; Kim, H.-J.; Park, J.-G.; Alfajaro, M.M.; Kim, D.-S.; Son, K.-Y.; Yang, D.-K.; Hyun, B.-H.; et al. Reassortment among bovine, porcine and human rotavirus strains results in G8P[7] and G6P[7] strains isolated from cattle in South Korea. Vet. Microbiol. 2011, 152, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Periz, J.; Celma, C.; Jing, B.; Pinkney, J.N.M.; Roy, P.; Kapanidis, A.N. Rotavirus mRNAS are released by transcript-specific channels in the double-layered viral capsid. Proc. Natl. Acad. Sci. USA 2013, 110, 12042–12047. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Wang, G.J.; Clerx, J.P.M.; Chiu, W. Three-dimensional structure of rotavirus. J. Mol. Biol. 1988, 199, 269–275. [Google Scholar] [CrossRef]
- Jiang, X.; Jayaram, H.; Kumar, M.; Ludtke, S.; Estes, M.K.; Prasad, B.V.V. Cryoelectron microscopy structures of rotavirus NSP2-NSP5 and NSP2-RNA complexes: Implications for genome replication. J. Virol. 2006, 80, 10829–10835. [Google Scholar] [CrossRef]
- Vasquez-Del Carpio, R.; Gonzalez-Nilo, F.D.; Riadi, G.; Taraporewala, Z.F.; Patton, J.T. Histidine triad-like motif of the rotavirus NSP2 octamer mediates both RTPase and NTPase activities. J. Mol. Biol. 2006, 362, 539–554. [Google Scholar] [CrossRef]
- Borodavka, A.; Dykeman, E.C.; Schrimpf, W.; Lamb, D.C. Protein-mediated RNA folding governs sequence-specific interactions between rotavirus genome segments. Elife 2017, 6, e27453. [Google Scholar] [CrossRef]
- Eichwald, C.; Rodriguez, J.F.; Burrone, O.R. Characterization of rotavirus NSP2/NSP5 interactions and the dynamics of viroplasm formation. J. Gen. Virol. 2004, 85, 625–634. [Google Scholar] [CrossRef]
- Buttafuoco, A.; Michaelsen, K.; Tobler, K.; Ackermann, M.; Fraefel, C.; Eichwald, C. Conserved Rotavirus NSP5 and VP2 Domains Interact and Affect Viroplasm. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Berkova, Z.; Crawford, S.E.; Trugnan, G.; Yoshimori, T.; Morris, A.P.; Estes, M.K. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J. Virol. 2006, 80, 6061–6071. [Google Scholar] [CrossRef]
- Patton, J.T.; Silvestri, L.S.; Tortorici, M.A.; Carpio, R.V.-D.; Taraporewala, Z.F. Rotavirus Genome Replication and Morphogenesis: Role of the Viroplasm. In Reoviruses: Entry, Assembly and Morphogenesis; Springer: New York, NY, USA, 2006; Volume 309, pp. 169–187. [Google Scholar] [CrossRef]
- Chang-Graham, A.L.; Perry, J.L.; Strtak, A.C.; Ramachandran, N.K.; Criglar, J.M.; Philip, A.A.; Patton, J.T.; Estes, M.K.; Hyser, J.M. Rotavirus Calcium Dysregulation Manifests as Dynamic Calcium Signaling in the Cytoplasm and Endoplasmic Reticulum. Sci. Rep. 2019, 9, 10822. [Google Scholar] [CrossRef]
- Diaz, Y.; Chemello, M.E.; Pena, F.; Aristimuno, O.C.; Zambrano, J.L.; Rojas, H.; Bartoli, F.; Salazar, L.; Chwetzoff, S.; Sapin, C.; et al. Expression of nonstructural rotavirus protein NSP4 mimics Ca2+ homeostasis changes induced by rotavirus infection in cultured cells. J. Virol. 2008, 82, 11331–11343. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Collinson-Pautz, M.R.; Utama, B.; Estes, M.K. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBio 2010, 1, e00265-10. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Utama, B.; Crawford, S.E.; Broughman, J.R.; Estes, M.K. Activation of the endoplasmic reticulum calcium sensor STIM1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J. Virol. 2013, 87, 13579–13588. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Estes, M.K.; Hu, Y.; Ball, J.M.; Zeng, C.Q.; Schilling, W.P. The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J. Virol. 1995, 69, 5763–5772. [Google Scholar] [CrossRef]
- Meyer, J.C.; Bergmann, C.C.; Bellamy, A.R. Interaction of rotavirus cores with the nonstructural glycoprotein NS28. Virology 1989, 171, 98–107. [Google Scholar] [CrossRef]
- Au, K.S.; Chan, W.K.; Burns, J.W.; Estes, M.K. Receptor activity of rotavirus nonstructural glycoprotein NS28. J. Virol. 1989, 63, 4553–4562. [Google Scholar] [CrossRef]
- Au, K.-S.; Mattion, N.M.; Estes, M.K. A subviral particle binding domain on the rotavirus nonstructural glycoprotein NS28. Virology 1993, 194, 665–673. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Maass, D.; Poruchynsky, M.S.; Atkinson, P.H.; Bellamy, A.R. Topology of the non-structural rotavirus receptor glycoprotein NS28 in the rough endoplasmic reticulum. EMBO J. 1989, 8, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Vende, P.; Piron, M.; Castagné, N.; Poncet, D. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J. Virol. 2000, 74, 7064–7071. [Google Scholar] [CrossRef]
- Piron, M.; Delaunay, T.; Grosclaude, J.; Poncet, D. Identification of the RNA-binding, dimerization, and eIF4GI-binding domains of rotavirus nonstructural protein NSP3. J. Virol. 1999, 73, 5411–5421. [Google Scholar] [CrossRef] [PubMed]
- Piron, M.; Vende, P.; Cohen, J.; Poncet, D. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 1998, 17, 5811–5821. [Google Scholar] [CrossRef] [PubMed]
- Poncet, D.; Laurent, S.; Cohen, J. Four nucleotides are the minimal requirement for RNA recognition by rotavirus non-structural protein NSP3. EMBO J. 1994, 13, 4165–4173. [Google Scholar] [CrossRef]
- Barro, M.; Patton, J.T. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. USA 2005, 102, 4114–4119. [Google Scholar] [CrossRef]
- Dunn, S.J.; Cross, T.L.; Greenberg, H.B. Comparison of the rotavirus nonstructural protein NSP1 (NS53) from different species by sequence analysis and northern blot hybridization. Virology 1994, 203, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Schnepf, N.; Deback, C.; Dehee, A.; Gault, E.; Parez, N.; Garbarg-Chenon, A. Rearrangements of rotavirus genomic segment 11 are generated during acute infection of immunocompetent children and do not occur at random. J. Virol. 2008, 82, 3689–3696. [Google Scholar] [CrossRef]
- Troupin, C.; Schnuriger, A.; Duponchel, S.; Deback, C.; Schnepf, N.; Dehee, A.; Garbarg-Chenon, A. Rotavirus rearranged genomic RNA segments are preferentially packaged into viruses despite not conferring selective growth advantage to viruses. PLoS ONE 2011, 6, e20080. [Google Scholar] [CrossRef]
- Patton, J.T.; Taraporewala, Z.; Chen, D.; Chizhikov, V.; Jones, M.; Elhelu, A.; Collins, M.; Kearney, K.; Wagner, M.; Hoshino, Y.; et al. Effect of intragenic rearrangement and changes in the 3′ consensus sequence on NSP1 expression and rotavirus replication. J. Virol. 2001, 75, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Heiman, E.M.; McDonald, S.M.; Barro, M.; Taraporewala, Z.F.; Bar-Magen, T.; Patton, J.T. Group A human rotavirus genomics: Evidence that gene constellations are influenced by viral protein interactions. J. Virol. 2008, 82, 11106–11116. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary dynamics of human rotaviruses: Balancing reassortment with preferred genome constellations. PLoS Pathog. 2009, 5, e1000634. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, S.; Yamada, Y.; Kinouchi, M.; Kudo, Y.; Ikemura, T. Codon usage and tRNA genes in eukaryotes: Correlation of codon usage diversity with translation efficiency and with CG-Dinucleotide usage as assessed by multivariate analysis. J. Mol. Evol. 2001, 53, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, W.J.; Peng, S.W.; Sun, X.Y.; Frazer, I. Papillomavirus capsid protein expression level depends on the match between codon usage and tRNA availability. J. Virol. 1999, 73, 4972–4982. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef]
- Mioduser, O.; Goz, E.; Tuller, T. Significant differences in terms of codon usage bias between bacteriophage early and late genes: A comparative genomics analysis. BMC Genom. 2017, 18, 866. [Google Scholar] [CrossRef]
- Miller, J.B.; Hippen, A.A.; Wright, S.M.; Morris, C.; Ridge, P.G. Human viruses have codon usage biases that match highly expressed proteins in the tissues they infect. Biomed. Genet. Genom. 2017. [Google Scholar] [CrossRef]
- Aragonès, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathog. 2010, 6, e1000797. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Singhal, S.; Kumar, U.; Ansari, A.; Tiwari, R.; Dhama, K.; Das, J.; Munjal, A.; Singh, R.K. Analysis of Nipah Virus Codon Usage and Adaptation to Hosts. Front. Microbiol. 2019, 10, 886. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Acevedo, A.; Cooper, S.B.; Andino, R. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 2012, 12, 623–632. [Google Scholar] [CrossRef]
- Luo, W.; Roy, A.; Guo, F.; Irwin, D.M.; Shen, X.; Pan, J.; Shen, Y. Host Adaptation and Evolutionary Analysis of Zaire ebolavirus: Insights From Codon Usage Based Investigations. Front. Microbiol. 2020, 11, 2823. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef]
- Hussain, S.; Shinu, P.; Islam, M.; Chohan, M.S.; Rasool, S.T. Analysis of Codon Usage and Nucleotide Bias in Middle East Respiratory Syndrome Coronavirus Genes. Evol. Bioinform. 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.-R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef]
- Bradel-Tretheway, B.G.; Zhen, Z.; Dewhurst, S. Effects of codon-optimization on protein expression by the human herpesvirus 6 and 7 U51 open reading frame. J. Virol. Methods 2003, 111, 145–156. [Google Scholar] [CrossRef]
- Ngumbela, K.C.; Ryan, K.P.; Sivamurthy, R.; Brockman, M.A.; Gandhi, R.T.; Bhardwaj, N.; Kavanagh, D.G. Quantitative effect of suboptimal codon usage on translational efficiency of mRNA encoding HIV-1 gag in intact T cells. PLoS ONE 2008, 3, e2356. [Google Scholar] [CrossRef]
- Kattoor, J.J.; Malik, Y.S.; Sasidharan, A.; Rajan, V.M.; Dhama, K.; Ghosh, S.; Banyai, K.; Kobayashi, N.; Singh, R.K. Analysis of co-don usage pattern evolution in avian rotaviruses and their preferred host. Infect. Genet. Evol. 2015, 34, 17–25. [Google Scholar] [CrossRef]
- Gómez, M.M.; Tort, L.F.L.; Volotao, E.D.M.; Recarey, R.; Moratorio, G.; Musto, H.; Leite, J.P.G.; Cristina, J. Analysis of human P[4]G2 rotavirus strains isolated in Brazil reveals codon usage bias and strong compositional constraints. Infect. Genet. Evol. 2011, 11, 580–586. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jitobaom, K.; Phakaratsakul, S.; Sirihongthong, T.; Chotewutmontri, S.; Suriyaphol, P.; Suptawiwat, O.; Auewarakul, P. Codon usage similarity between viral and some host genes suggests a codon-specific translational regulation. Heliyon 2020, 6, e03915. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using time-structured data to estimate evolu-tionary rates of double-stranded DNA viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Holmes, E.C. What is the time-scale of hantavirus evolution? Infect. Genet. Evol. 2014, 25, 144–145. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Fourment, M.; Kosakovsky Pond, S.L. Inconsistencies in Estimating the Age of HIV-1 Subtypes Due to Heterotachy. Mol. Biol. Evol. 2012, 29, 451–456. [Google Scholar] [CrossRef]
- Duchêne, S.; Holmes, E.; Ho, S.Y.W. Analyses of evolutionary dynamics in viruses are hindered by a time-dependent bias in rate estimates. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140732. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef]
- Wang, T.H.; Donaldson, Y.K.; Brettle, R.P.; Bell, J.E.; Simmonds, P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 2001, 75, 11686–11699. [Google Scholar] [CrossRef]
- Slatkin, M.; Maddison, W.P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 1989, 123, 603–613. [Google Scholar] [CrossRef]
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef]
- Puigbò, P.; Romeu, A.; Garcia-Vallve, S. HEG-DB: A database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res. 2007, 36, D524–D527. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallvé, S. E-CAI: A novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinform. 2008, 9, 1–7. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Mingo, R.; Zhang, S.; Long, C.P.; LaConte, L.E.W.; McDonald, S.M. Genetic determinants restricting the reassortment of heterologous NSP2 genes into the simian rotavirus SA11 genome. Sci. Rep. 2017, 7, 9301. [Google Scholar] [CrossRef] [PubMed]
- Rabadan, R.; Levine, A.J.; Krasnitz, M. Non-random reassortment in human influenza A viruses. Influenza Other Respir. Viruses 2008, 2, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Selective Genome Packaging Mechanisms of Influenza A Viruses. Cold Spring Harb. Perspect. Med. 2021, 11, a038497. [Google Scholar] [CrossRef]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef]
- McDonald, S.M.; Patton, J.T. Assortment and packaging of the segmented rotavirus genome. Trends Microbiol. 2011, 19, 136–144. [Google Scholar] [CrossRef]
- Suzuki, Y. A candidate packaging signal of human rotavirus differentiating Wa-like and DS-1-like genomic constellations. Microbiol. Immunol. 2015, 59, 567–571. [Google Scholar] [CrossRef]
- Li, W.; Manktelow, E.; von Kirchbach, J.C.; Gog, J.R.; Desselberger, U.; Lever, A.M. Genomic analysis of codon, sequence and structural conservation with selective biochemical-structure mapping reveals highly conserved and dynamic structures in rotavirus RNAs with potential cis-acting functions. Nucleic Acids Res. 2010, 38, 7718–7735. [Google Scholar] [CrossRef]
- Fajardo, T., Jr.; Sung, P.-Y.; Celma, C.C.; Roy, P. Rotavirus Genomic RNA Complex Forms via Specific RNA–RNA Interactions: Disruption of RNA Complex Inhibits Virus Infectivity. Viruses 2017, 9, 167. [Google Scholar] [CrossRef]
- Borodavka, A.; Desselberger, U.; Patton, J.T. Genome packaging in multi-segmented dsRNA viruses: Distinct mechanisms with similar outcomes. Curr. Opin. Virol. 2018, 33, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Long, C.P.; McDonald, S.M. Rotavirus genome replication: Some assembly required. PLoS Pathog. 2017, 13, e1006242. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Nuyts, V.; Heylen, E.; De Coster, S.; Conceição-Neto, N.; Van Ranst, M.; Matthijnssens, J. Emergence of human G2P[4] rotaviruses containing animal derived gene segments in the post-vaccine era. Sci. Rep. 2016, 6, 36841. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Kanai, Y.; Fukuda, S.; Kugita, M.; Kawagishi, T.; Ito, N.; Sugiyama, M.; Matsuura, Y.; Kobayashi, T.; Taniguchi, K. Reverse Genetics System Demonstrates that Rotavirus Nonstructural Protein NSP6 Is Not Essential for Viral Replication in Cell Culture. J. Virol. 2017, 91, e00695-17. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Li, W.; Manktelow, E.; Lever, J.; Easton, L.E.; Lukavsky, P.J.; Desselberger, U.; Lever, A.M. Physicochemical analysis of rotavirus segment 11 supports a ’modified panhandle’ structure and not the predicted alternative tRNA-like structure (TRLS). Arch. Virol. 2014, 159, 235–248. [Google Scholar] [CrossRef][Green Version]
- Hu, L.; Sankaran, B.; Laucirica, D.R.; Patil, K.; Salmen, W.; Ferreon, A.C.M.; Tsoi, P.S.; Lasanajak, Y.; Smith, D.F.; Ramani, S.; et al. Glycan recognition in globally dominant human rotaviruses. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Xia, M.; Tan, M.; Zhong, W.; Wei, C.; Wang, L.; Morrow, A.; Jiang, X. Spike protein VP8* of human rotavirus recognizes histo-blood group antigens in a type-specific manner. J. Virol. 2012, 86, 4833–4843. [Google Scholar] [CrossRef]
- Arias, C.F.; Silva-Ayala, D.; López, S. Rotavirus entry: A deep journey into the cell with several exits. J. Virol. 2015, 89, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Prim. 2017, 3, 17083. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Yinda, C.K.; Zeller, M.; Conceição-Neto, N.; Maes, P.; Deboutte, W.; Beller, L.; Heylen, E.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent reassortant bat rotaviruses in Cameroon, without evidence of zoonosis. Sci. Rep. 2016, 6, 34209. [Google Scholar] [CrossRef]
- Santos, N.; Hoshino, Y. Global distribution of rotavirus serotypes/genotypes and its implication for the development and implementation of an effective rotavirus vaccine. Rev. Med. Virol. 2005, 15, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Hoxie, I.; Dennehy, J.J. Intragenic recombination influences rotavirus diversity and evolution. Virus Evol. 2020, 6. [Google Scholar] [CrossRef]
- Altmann, M.; Müller, P.P.; Pelletier, J.; Sonenberg, N.; Trachsel, H. A mammalian translation initiation factor can substitute for its yeast homologue in vivo. J. Biol. Chem. 1989, 264, 12145–12147. [Google Scholar] [CrossRef]
- Barro, M.; Patton, J.T. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 2007, 81, 4473–4481. [Google Scholar] [CrossRef]
- Bagchi, P.; Dutta, D.; Chattopadhyay, S.; Mukherjee, A.; Halder, U.C.; Sarkar, S.; Kobayashi, N.; Komoto, S.; Taniguchi, K.; Chawla-Sarkar, M. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J. Virol. 2010, 84, 6834–6845. [Google Scholar] [CrossRef]
- Bagchi, P.; Nandi, S.; Nayak, M.K.; Chawla-Sarkar, M. Molecular mechanism behind rotavirus NSP1-mediated PI3 kinase activation: Interaction between NSP1 and the p85 subunit of PI3 kinase. J. Virol. 2012, 87, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.W.; Ettayebi, K.; Hardy, M.E. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: A novel mechanism of IFN antagonism. PLoS Pathog. 2009, 5, e1000280. [Google Scholar] [CrossRef]
- Motayo, B.; Oluwasemowo, O.O.; Olusola, B.A.; Opayele, A.V.; Faneye, A. Phylogeography and evolutionary analysis of African Rotavirus a genotype G12 reveals district genetic diversification within lineage III. Heliyon 2019, 5, e02680. [Google Scholar] [CrossRef]
- Arana, A.; Jere, K.C.; Chaguza, C.; Montes, M.; Alkorta, M.; Iturriza-Gomara, M.; Cilla, G. Molecular epidemiology of G12 ro-tavirus strains during eight consecutive epidemic seasons in the Basque Country (North of Spain), 2010–2018. Infect. Genet. Evol. 2019, 71, 67–75. [Google Scholar] [CrossRef]
- Zeller, M.; Heylen, E.; Damanka, S.; Pietsch, C.; Donato, C.; Tamura, T.; Kulkarni, R.; Arora, R.; Cunliffe, N.; Maunula, L.; et al. Emerging OP354-Like P[8] Rotaviruses Have Rapidly Dispersed from Asia to Other Continents. Mol. Biol. Evol. 2015, 32, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.E.; Mutuc, J.D.; Panozzo, C.A.; Payne, D.C.; Cortese, M.M.; Cortes, J.E.; Yen, C.; Esposito, D.H.; Lopman, B.A.; Patel, M.M.; et al. Sustained decline in rotavirus detections in the United States following the introduction of rotavirus vaccine in 2006. Pediatr. Infect. Dis. J. 2011, 30, S30–S34. [Google Scholar] [CrossRef] [PubMed]
- Almalki, S.S.R. Circulating rotavirus G and P strains post rotavirus vaccination in Eastern Mediterranean Region. Saudi Med. J. 2018, 39, 755–766. [Google Scholar] [CrossRef]
- Ivashechkin, A.A.; Yuzhakov, A.G.; Grebennikova, T.V.; Yuzhakova, K.A.; Kulikova, N.; Kisteneva, L.B.; Smetanina, S.V.; Bazarova, M.V.; Almazova, M.G. Genetic diversity of group A rotaviruses in Moscow in 2018-2019. Arch. Virol. 2020, 165, 691–702. [Google Scholar] [CrossRef]
- Damtie, D.; Melku, M.; Tessema, B.; Vlasova, A.N. Prevalence and Genetic Diversity of Rotaviruses among under-Five Children in Ethiopia: A Systematic Review and Meta-Analysis. Viruses 2020, 12, 62. [Google Scholar] [CrossRef]
- Hoque, S.A.; Khandoker, N.; Thongprachum, A.; Khamrin, P.; Takanashi, S.; Okitsu, S.; Nishimura, S.; Kikuta, H.; Yamamoto, A.; Sugita, K.; et al. Distribution of rotavirus gen-otypes in Japan from 2015 to 2018: Diversity in genotypes before and after introduction of rotavirus vaccines. Vaccine 2020, 38, 3980–3986. [Google Scholar] [CrossRef]
- Jere, K.C.; Bar-Zeev, N.; Chande, A.; Bennett, A.; Pollock, L.; Sanchez-Lopez, P.F.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Vaccine Effectiveness against DS-1-Like Rotavirus Strains in Infants with Acute Gastroenteritis, Malawi, 2013–2015. Emerg. Infect. Dis. 2019, 25, 1734–1737. [Google Scholar] [CrossRef]
- Nomikou, K.; Hughes, J.; Wash, R.; Kellam, P.; Breard, E.; Zientara, S.; Palmarini, M.; Biek, R.; Mertens, P. Widespread Reassortment Shapes the Evolution and Epidemiology of Bluetongue Virus following European Invasion. PLoS Pathog. 2015, 11, e1005056. [Google Scholar] [CrossRef]
- Jacquot, M.; Rao, P.P.; Yadav, S.; Nomikou, K.; Maan, S.; Jyothi, Y.K.; Reddy, N.; Putty, K.; Hemadri, D.; Singh, K.P.; et al. Contrasting selective patterns across the segmented genome of bluetongue virus in a global reassortment hotspot. Virus Evol. 2019, 5, vez027. [Google Scholar] [CrossRef]
- Samy, A.; Peterson, A.T.; Hall, M. Phylogeography of Rift Valley Fever Virus in Africa and the Arabian Peninsula. PLoS Negl. Trop. Dis. 2017, 11, e0005226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, L.; Du, J.; Hu, X.; Xie, Y.; Wu, J.; Lin, X.; Yin, N.; Sun, M.; Li, H. MicroRNA-7 Inhibits Rotavirus Replication by Targeting Viral NSP5 in vivo and in vitro. Viruses 2020, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- López, T.; Rojas, M.; Ayala-Bretón, C.; Lopez, S.; Arias, C.F. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. J. Gen. Virol. 2005, 86, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Contin, R.; Arnoldi, F.; Campagna, M.; Burrone, O.R. Rotavirus NSP5 orchestrates recruitment of viroplasmic proteins. J. Gen. Virol. 2010, 91, 1782–1793. [Google Scholar] [CrossRef]
- Rabadan, R.; Levine, A.J.; Robins, H. Comparison of avian and human influenza A viruses reveals a mutational bias on the viral genomes. J. Virol. 2006, 80, 589–600. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Association Index (AI) | p-Value | Parsimony Score (PS) | p-Value |
|---|---|---|---|---|
| Segment 1 | 1.79 | 0.00 | 16.78 | 0.00 |
| Segment 2 | 1.97 | 0.00 | 18.63 | 0.00 |
| Segment 3 | 2.88 | 0.00 | 22.86 | 0.00 |
| Segment 4 | 1.23 | 0.00 | 11.71 | 0.00 |
| Segment 5 | 2.11 | 0.00 | 17.87 | 0.00 |
| Segment 6 | 2.68 | 0.00 | 19.19 | 0.00 |
| Segment 7 | 1.60 | 0.00 | 18.03 | 0.00 |
| Segment 8 | 1.89 | 0.00 | 19.66 | 0.00 |
| Segment 9 | 3.83 | 0.00 | 25.79 | 0.00 |
| Segment 10 | 2.44 | 0.00 | 17.26 | 0.00 |
| Segment 11 | 2.11 | 0.00 | 18.99 | 0.00 |
| Segment 4 * | 3.11 | 0.00 | 31.88 | 0.00 |
| Segment 9 * | 4.18 | 0.00 | 34.15 | 0.00 |
| Segment | Mean Rate | 95% HPD (Lower, Upper) | Coefficient of Variation | TMRCA | |
|---|---|---|---|---|---|
| Segment 1 (VP1) | 1.724 × 10−3 | 1.610 × 10−3 | 1.835 × 10−3 | 2.410 | 1957 |
| Segment 2 (VP2) | 1.703 × 10−3 | 1.589 × 10−3 | 1.826 × 10−3 | 2.602 | 1963 |
| Segment 3 (VP3) | 1.948 × 10−3 | 1.814 × 10−3 | 2.087 × 10−3 | 2.436 | 1962 |
| Segment 4 (VP4) | 3.775 × 10−3 | 3.517 × 10−3 | 4.044 × 10−3 | 8.978 | 1964.5 |
| Segment 5 (NSP1) | 2.953 × 10−3 | 2.694 × 10−3 | 3.223 × 10−3 | 3.994 | 1964.5 |
| Segment 6 (VP6) | 1.694 × 10−3 | 1.541 × 10−3 | 1.857 × 10−3 | 3.412 | 1967.5 |
| Segment 7 (NSP3) | 1.615 × 10−3 | 1.454 × 10−3 | 1.778 × 10−3 | 5.061 | 1967 |
| Segment 8 (NSP2) | 1.473 × 10−3 | 1.335 × 10−3 | 1.610 × 10−3 | 3.323 | 1966.5 |
| Segment 9 (VP7) | 2.660 × 10−3 | 2.400 × 10−3 | 2.940 × 10−3 | 3.375 | 1966.5 |
| Segment 10 (NSP4) | 1.583 × 10−3 | 1.420 × 10−3 | 1.743 × 10−3 | 2.599 | 1967.5 |
| Segment 11 (NSP5/6) | 2.424 × 10−3 | 2.040 × 10−3 | 2.831 × 10−3 | 3.285 | 1969 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoxie, I.; Dennehy, J.J. Rotavirus A Genome Segments Show Distinct Segregation and Codon Usage Patterns. Viruses 2021, 13, 1460. https://doi.org/10.3390/v13081460
Hoxie I, Dennehy JJ. Rotavirus A Genome Segments Show Distinct Segregation and Codon Usage Patterns. Viruses. 2021; 13(8):1460. https://doi.org/10.3390/v13081460
Chicago/Turabian StyleHoxie, Irene, and John J. Dennehy. 2021. "Rotavirus A Genome Segments Show Distinct Segregation and Codon Usage Patterns" Viruses 13, no. 8: 1460. https://doi.org/10.3390/v13081460
APA StyleHoxie, I., & Dennehy, J. J. (2021). Rotavirus A Genome Segments Show Distinct Segregation and Codon Usage Patterns. Viruses, 13(8), 1460. https://doi.org/10.3390/v13081460