
A Diverse Viral Community from Predatory Wasps in Their Native and Invaded Range, with a New Virus Infectious to Honey Bees

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Vespula vulgaris Collections, RNA Extractions and RNA Sequencing
2.2. Identification and Phylogenetic Analysis of Novel Virus Sequences
2.3. PCR Validation of Novel Viruses
2.4. Virus Propagation and Screening in Honey Bees
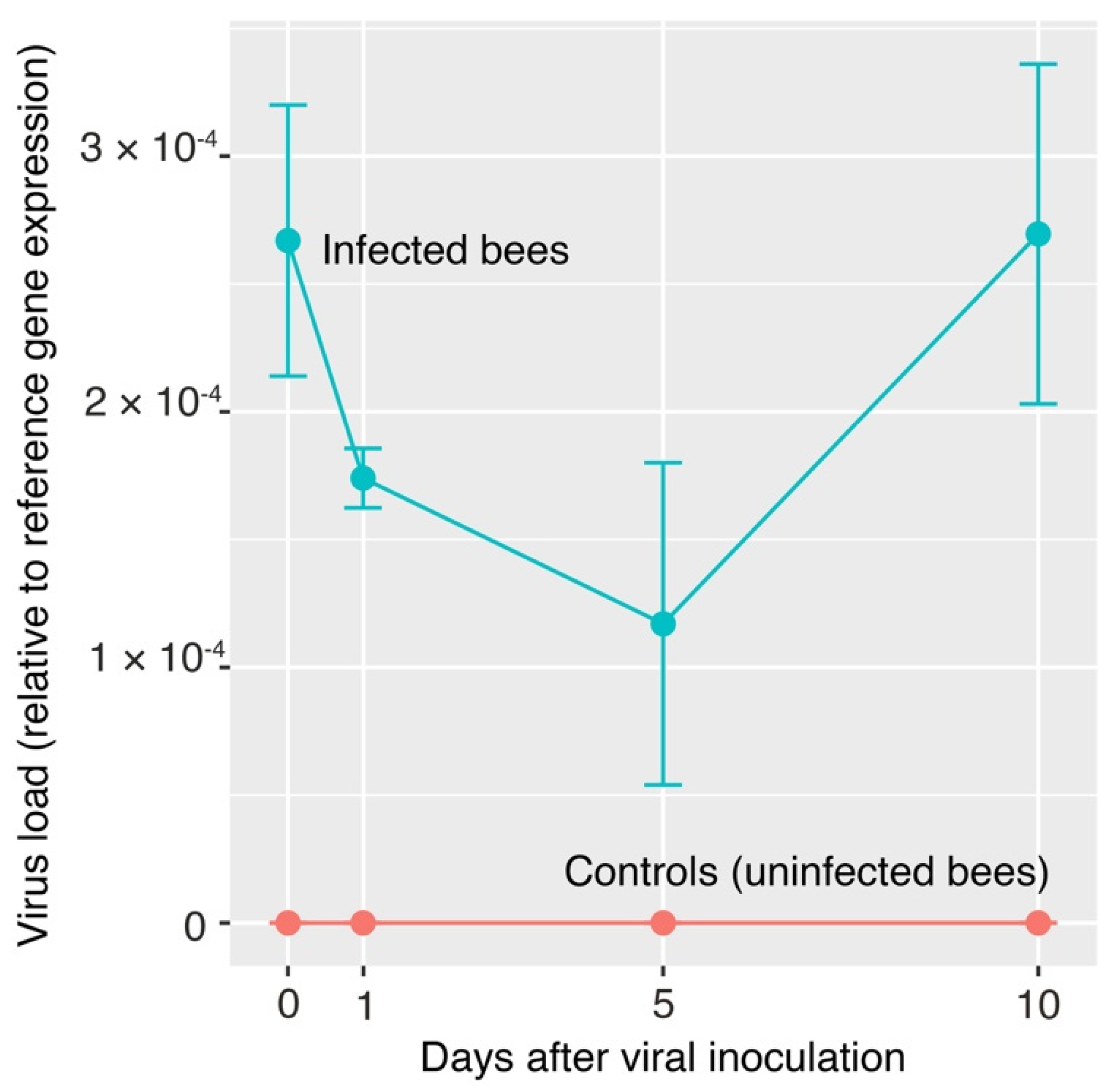
2.5. Moku-Like Viral Load in Inoculated Honey Bees
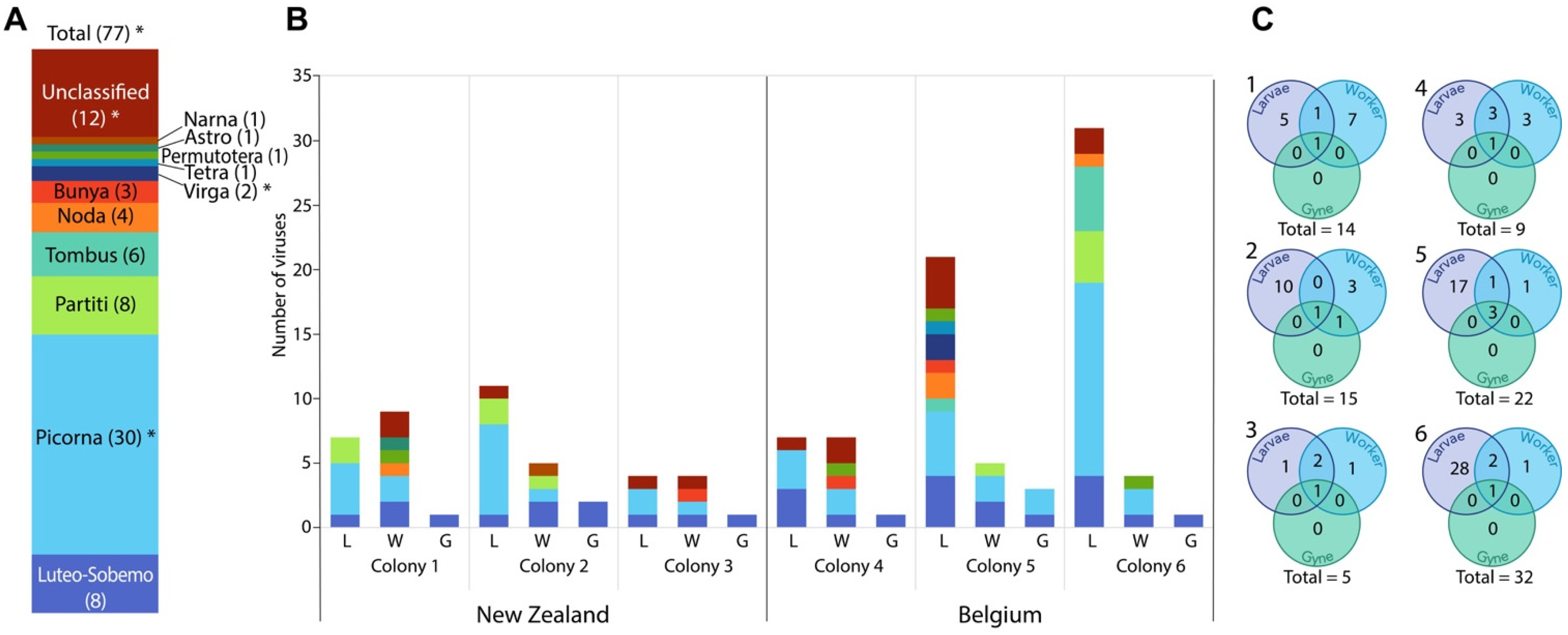
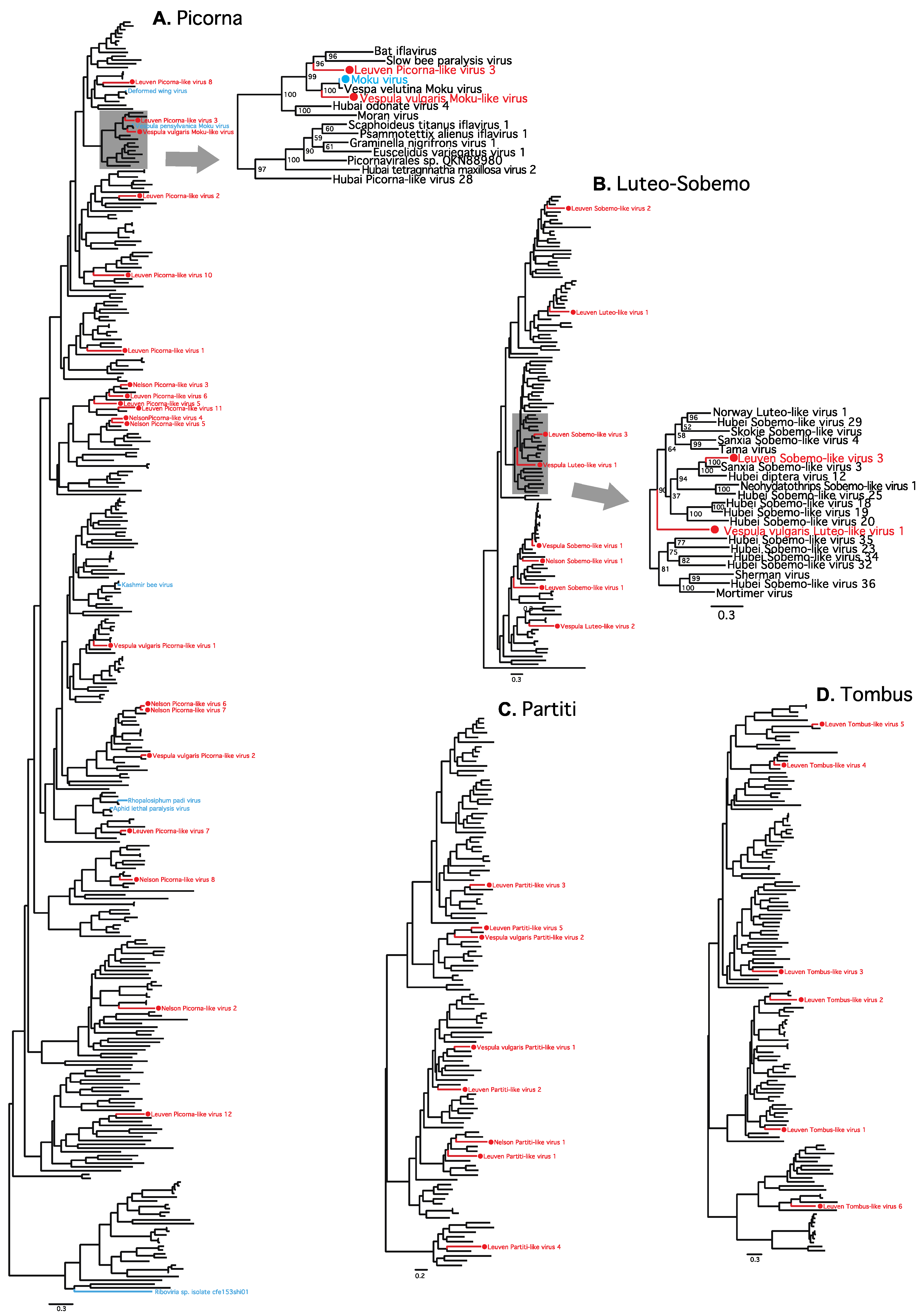
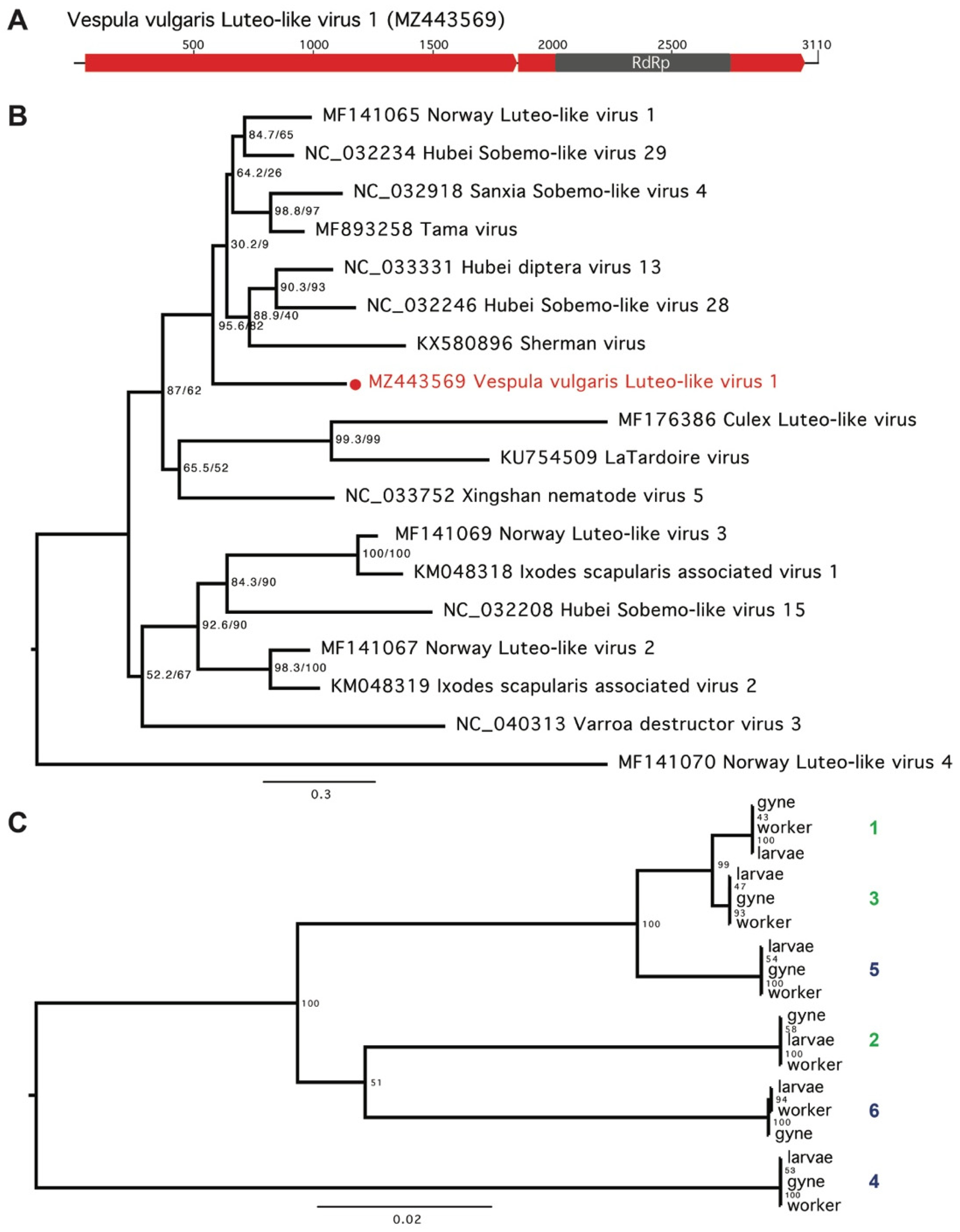
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J.; et al. Abundant and diverse RNA viruses in insects revealed by RNA-seq analysis: Ecological and evolutionary implications. mSystems 2020, 5, e00039-20. [Google Scholar] [CrossRef]
- De Miranda, J.; Cornman, R.; Evans, J.; Semberg, E.; Haddad, N.; Neumann, P.; Gauthier, L. Genome characterization, prevalence and distribution of a macula-like virus from Apis mellifera and Varroa destructor. Viruses 2015, 7, 2789. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, D.A.; Fuller, Z.L.; Ray, A.M.; Brockmann, A.; Frazier, M.; Gikungu, M.W.; Martinez, J.F.I.; Kapheim, K.M.; Kerby, J.T.; Kocher, S.D.; et al. Investigating the viral ecology of global bee communities with high-throughput metagenomics. Sci. Rep. 2018, 8, 8879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordecai, G.J.; Brettell, L.E.; Pachori, P.; Villalobos, E.M.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Moku virus; a new iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 2016, 6, 34983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.K.; Anderson, D.L.; Durr, P.A. Metagenomic analysis of Varroa-free Australian honey bees (Apis mellifera) shows a diverse picornavirales virome. J. Gen. Virol. 2018, 99, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Remnant, E.J.; Shi, M.; Buchmann, G.; Blaquiere, T.; Holmes, E.C.; Beekman, M.; Ashe, A. A diverse range of novel RNA viruses in geographically distinct honey bee populations. J. Virol. 2017, 91, e00158-17. [Google Scholar] [CrossRef] [Green Version]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.-C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457. [Google Scholar] [CrossRef] [Green Version]
- Plowright, R.K.; Parrish, C.R.; McCallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Manley, R.; Boots, M.; Wilfert, L. Emerging viral disease risk to pollinating insects: Ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 2015, 52, 331–340. [Google Scholar] [CrossRef]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; vanEngelsdorp, D.; Lipkin, W.I.; dePamphillis, C.W.; Toth, A.L.; Cox-Foster, D.L. Rna viruses in hymenopteran pollinators: Evidence of inter-taxa virus transmission via pollen and potential impact on non-apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef]
- McMahon, D.P.; Wilfert, L.; Paxton, R.J.; Brown, M.J.F. Emerging viruses in bees: From molecules to ecology. Adv. Virus Res. 2018, 101, 251–291. [Google Scholar]
- Tehel, A.; Brown, M.J.; Paxton, R.J. Impact of managed honey bee viruses on wild bees. Curr. Opin. Virol. 2016, 19, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.J.; Highfield, A.C.; Brettell, L.; Villalobos, E.M.; Budge, G.E.; Powell, M.; Nikaido, S.; Schroeder, D. Global honey bee viral landscape altered by a parasitic mite. Science 2012, 336, 1304–1306. [Google Scholar] [CrossRef]
- Wilfert, L.; Long, G.; Leggett, H.C.; Schmid-Hempel, P.; Butlin, R.; Martin, S.J.M.; Boots, M. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 2016, 351, 594–597. [Google Scholar] [CrossRef] [Green Version]
- Mondet, F.; de Miranda, J.R.; Kretzschmar, A.; Le Conte, Y.; Mercer, A.R. On the front line: Quantitative virus dynamics in honeybee (Apis mellifera L.) colonies along a new expansion front of the parasite Varroa destructor. PLoS Pathog. 2014, 10, e1004323. [Google Scholar] [CrossRef] [Green Version]
- Evison, S.E.F.; Roberts, K.E.; Laurenson, L.; Pietravalle, S.; Hui, J.; Biesmeijer, J.C.; Smith, J.E.; Budge, G.; Hughes, W.O.H. Pervasiveness of parasites in pollinators. PLoS ONE 2012, 7, e30641. [Google Scholar] [CrossRef] [Green Version]
- Loope, K.J.; Baty, J.W.; Lester, P.J.; Wilson Rankin, E.E. Pathogen shifts in a honeybee predator following the arrival of the Varroa mite. Proc. R. Soc. B 2019, 286, 20182499. [Google Scholar] [CrossRef] [Green Version]
- Brettell, L.E.; Schroeder, D.C.; Martin, S.J. RNAseq analysis reveals virus diversity within Hawaiian apiary insect communities. Viruses 2019, 11, 397. [Google Scholar] [CrossRef] [Green Version]
- Dobelmann, J.; Felden, A.; Lester, P.J. Genetic strain diversity of multi-host RNA viruses that infect a wide range of pollinators and associates is shaped by geographic origins. Viruses 2020, 12, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.G.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef]
- Sébastien, A.; Lester, P.J.; Hall, R.J.; Wang, J.; Moore, N.E.; Gruber, M.A.M. Invasive ants carry novel viruses in their new range and form reservoirs for a honeybee pathogen. Biol. Lett. 2015, 11, 20150610. [Google Scholar] [CrossRef]
- Gruber, M.A.M.; Cooling, M.; Baty, J.W.; Buckley, K.; Friedlander, A.; Quinn, O.; Russell, J.F.E.J.; Sébastien, A.; Lester, P.J. Single-stranded RNA viruses infecting the invasive argentine ant, Linepithema humile. Sci. Rep. 2017, 7, 3304. [Google Scholar] [CrossRef] [Green Version]
- Archer, M.E. Vespine Wasps of the World: Behaviour, Ecology and Taxonomy of the Vespinae; Siri Sci. Press: Manchester, UK, 2012. [Google Scholar]
- Lester, P.J.; Beggs, J.R. Invasion success and management strategies for social Vespula wasps. Ann. Rev. Entomol. 2019, 64, 51–71. [Google Scholar] [CrossRef] [PubMed]
- Harrop, T.W.R.; Guhlin, J.; McLaughlin, G.M.; Permina, E.; Stockwell, P.; Gilligan, J.; Le Lec, M.F.; Gruber, M.A.M.; Quinn, O.; Lovegrove, M.; et al. High-quality assemblies for three invasive social wasps from the Vespula genus. G3 2020, 10, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.R. Social wasp (Hymenoptera: Vespidae) foraging behavior. Ann. Rev. Entomol. 2000, 45, 121–150. [Google Scholar] [CrossRef]
- Clapperton, B.K.; Alspach, P.A.; Moller, H.; Matheson, A.G. The impact of common and german wasps (Hymenoptera: Vespidae) on the new zealand beekeeping industry. N. Z. J. Zool. 1989, 16, 325–332. [Google Scholar] [CrossRef]
- Wilson, E.E.; Holway, D.A. Multiple mechanisms underlie displacement of solitary Hhawaiian Hymenoptera by an invasive social wasp. Ecology 2010, 91, 3294–3302. [Google Scholar] [CrossRef] [Green Version]
- Quinn, O.; Gruber, M.A.M.; Brown, R.L.; Baty, J.W.; Bulgarella, M.; Lester, P.J. A metatranscriptomic analysis of diseased social wasps (Vespula vulgaris) for pathogens, with an experimental infection of larvae and nests. PLoS ONE 2019, 13, e0209589. [Google Scholar] [CrossRef]
- Gruber, M.A.M.; Quinn, O.; Baty, J.W.; Dobelmann, J.; Haywood, J.; Wenseleers, T.; Lester, P.J. Fitness and microbial networks of the common wasp, Vespula vulgaris (Hymenoptera: Vespidae), in its native and introduced ranges. Ecol. Entomol. 2019. [Google Scholar] [CrossRef]
- Brenton-Rule, E.C.; Dobelmann, J.; Baty, J.W.; Brown, R.L.; Dvorak, L.; Grangier, J.; Masciocchi, M.; McGrannachan, C.; Shortall, C.R.; Schmack, J.; et al. The origins of global invasions of the German wasp (Vespula germanica) and its infection with four honey bee viruses. Biol. Invasions 2018, 20, 3445–3460. [Google Scholar] [CrossRef]
- Loope, K.J.; Wilson Rankin, E.E. Viral load, not food availability or temperature, predicts colony longevity in an invasive eusocial wasp with plastic life history. Sci. Rep. 2021, 11, 10087. [Google Scholar] [CrossRef]
- Lester, P.J.; Bosch, P.J.; Gruber, M.A.M.; Kapp, E.A.; Peng, L.; Brenton-Rule, E.C.; Buchanan, J.; Stanislawek, W.L.; Archer, M.; Corley, J.C.; et al. No evidence of enemy release in pathogen and microbial communities of common wasps (Vespula vulgaris) in their native and introduced range. PLoS ONE 2015, 10, e0121358. [Google Scholar] [CrossRef]
- Garigliany, M.; Taminiau, B.; El Agrebi, N.; Cadar, D.; Gilliaux, G.; Hue, M.; Desmecht, D.; Daube, G.; Linden, A.; Farnir, F.; et al. Moku virus in invasive asian hornets, Belgium, 2016. Emerg. Infect. Dis. 2017, 23, 2109–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmon, A.; Gayral, P.; Decante, D.; Klopp, C.; Bigot, D.; Thomasson, M.; Herniou, E.A.; Alaux, C.; Le Conte, Y. Viruses in the invasive hornet Vespa velutina. Viruses 2019, 11, 1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Highfield, A.; Kevill, J.; Mordecai, G.; Hunt, J.; Henderson, S.; Sauvard, D.; Feltwell, J.; Martin, S.J.; Sumner, S.; Schroeder, D.C. Detection and replication of Moku virus in honey bees and social wasps. Viruses 2020, 12, 607. [Google Scholar] [CrossRef]
- Felden, A.; Baty, J.W.; Bulgarella, M.; Brown, R.L.; Dobelmann, J.; Gruber, M.A.M.; Quinn, O.; Lester, P.J. Viral and fungal pathogens associated with Pneumolaelaps niutirani (Acari: Laelapidae): A mite found in diseased nests of Vespula wasps. Insectes Sociaux 2020, 67, 83–93. [Google Scholar] [CrossRef]
- Quinn, O. Microbiota of an Invasive Wasp Vespula vulgaris and Hymenopteran Relatives; Victoria University of Wellington: Wellington, New Zealand; Available online: https://researcharchive.vuw.ac.nz/xmlui/handle/10063/6978?show=full (accessed on 13 April 2018).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. Modelfinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. Ufboot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of Phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, C.; Genersch, E. RT-PCR analysis of Deformed wing virus in honeybees (Apis mellifera) and mites (Varroa destructor). J. Gen. Virol. 2005, 86, 3419–3424. [Google Scholar] [CrossRef] [PubMed]
- Runckel, C.; Flenniken, M.L.; Engel, J.C.; Ruby, J.G.; Ganem, D.; Andino, R.; DeRisi, J.L. Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS ONE 2011. [Google Scholar] [CrossRef] [Green Version]
- De Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.E.; Chejanovsky, N.; Chen, Y.-P.; Gauthier, L.; Genersch, E.; de Graaf, D.C.; et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 2013, 52, 1–56. [Google Scholar] [CrossRef] [Green Version]
- Human, H.; Brodschneider, R.; Dietemann, V.; Dively, G.; Ellis, J.D.; Forsgren, E.; Fries, I.; Hatjina, F.; Hu, F.-L.; Jaffé, R.; et al. Miscellaneous standard methods for Apis mellifera research. J. Apic. Res. 2013, 52, 1–53. [Google Scholar] [CrossRef] [Green Version]
- Cameron, R.C.; Duncan, E.J.; Dearden, P.K. Stable reference genes for the measurement of transcript abundance during larval caste development in the honeybee. Apidologie 2013, 44, 357–366. [Google Scholar] [CrossRef]
- Baty, J.W.; Bulgarella, M.; Dobelmann, J.; Felden, A.; Lester, P.J. Viruses and their effects in ants (Hymenoptera:Formicidae). Myrmecol. News 2020, 30, 213–228. [Google Scholar]
- Martin, S.J.; Brettell, L.E. Deformed wing virus in honeybees and other insects. Annu. Rev. Virol. 2019, 6, 49–69. [Google Scholar] [CrossRef]
- Harris, R.J. Diet of the wasps Vespula vulgaris and V. Germanica in honeydew beech forest of the south island, New Zealand. N. Z. J. Zool. 1991, 18, 159–169. [Google Scholar] [CrossRef]
- Spradbery, J.P. Wasps: An Account of the Biology and Natural History of Social and Solitary Wasps, 2nd ed.; Packhard Publishing Ltd.: Chichester, UK, 1984; p. 424. [Google Scholar]
- Santoro, D.; Hartley, S.; Lester, P.J. Behaviourally specialized foragers are less efficient and live shorter lives than generalists in wasp colonies. Sci. Rep. 2019, 9, 5366. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, J.H.O.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from Northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [Green Version]
- Imrie, R.M.; Roberts, K.E.; Longdon, B. Between virus correlations in the outcome of infection across host species: Evidence of virus by host species interactions. Evol. Lett. 2021. [Google Scholar] [CrossRef]
- Rose, E.A.F.; Harris, R.J.; Glare, T.R. Possible pathogens of social wasps (Hymenoptera: Vespidae) and their potential as biological control agents. N. Z. J. Zool. 1999, 26, 179–190. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remnant, E.J.; Baty, J.W.; Bulgarella, M.; Dobelmann, J.; Quinn, O.; Gruber, M.A.M.; Lester, P.J. A Diverse Viral Community from Predatory Wasps in Their Native and Invaded Range, with a New Virus Infectious to Honey Bees. Viruses 2021, 13, 1431. https://doi.org/10.3390/v13081431
Remnant EJ, Baty JW, Bulgarella M, Dobelmann J, Quinn O, Gruber MAM, Lester PJ. A Diverse Viral Community from Predatory Wasps in Their Native and Invaded Range, with a New Virus Infectious to Honey Bees. Viruses. 2021; 13(8):1431. https://doi.org/10.3390/v13081431
Chicago/Turabian StyleRemnant, Emily J., James W. Baty, Mariana Bulgarella, Jana Dobelmann, Oliver Quinn, Monica A. M. Gruber, and Philip J. Lester. 2021. "A Diverse Viral Community from Predatory Wasps in Their Native and Invaded Range, with a New Virus Infectious to Honey Bees" Viruses 13, no. 8: 1431. https://doi.org/10.3390/v13081431
APA StyleRemnant, E. J., Baty, J. W., Bulgarella, M., Dobelmann, J., Quinn, O., Gruber, M. A. M., & Lester, P. J. (2021). A Diverse Viral Community from Predatory Wasps in Their Native and Invaded Range, with a New Virus Infectious to Honey Bees. Viruses, 13(8), 1431. https://doi.org/10.3390/v13081431