
Broad-Based Influenza-Specific CD8+ T Cell Response without the Typical Immunodominance Hierarchy and Its Potential Implication


Abstract
1. Introduction
2. Materials and Methods
2.1. Human Peripheral Blood Mononuclear Cells (PBMCs) and Antigen Presenting Cells (APCs)
2.2. Culture Medium, Synthetic Peptides, Antibodies and Viruses
2.3. Generation of Poly-Specificity CD8+ T Cell Lines In Vitro
2.4. rVV Infection for Identifying Protein-Specific CD8+ T Cells
2.5. Identification of Novel IAV-Specific CD8+ T Cell Epitopes and Its Minimal Epitopes
2.6. HLA Restriction Assay
2.7. Intracellular Cytokine Staining (ICS) and Antigen Presentation Kinetics Assay
3. Results
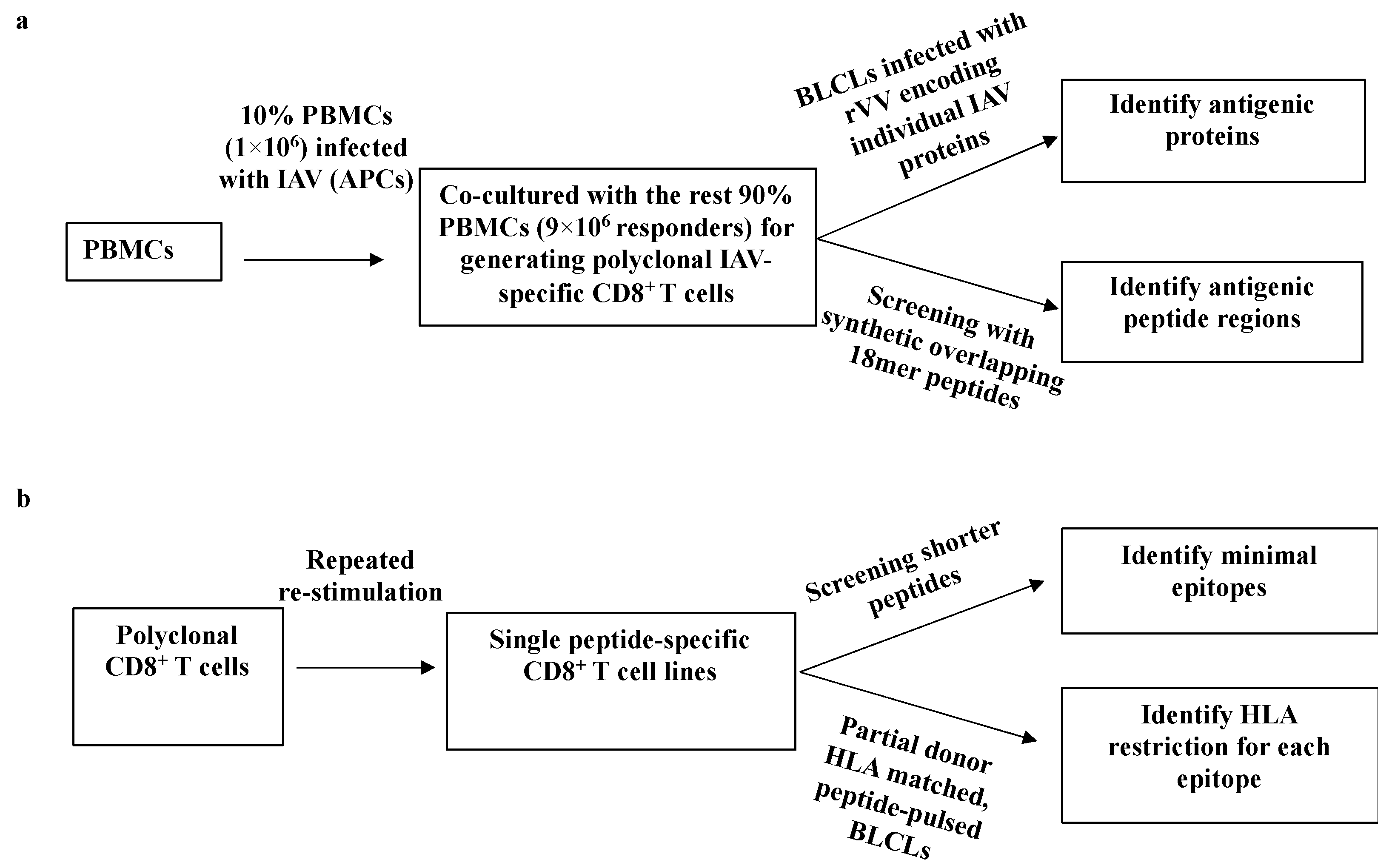
3.1. A Systematic Approach for Identifying IAV-Specific CD8+ T Cell Responses
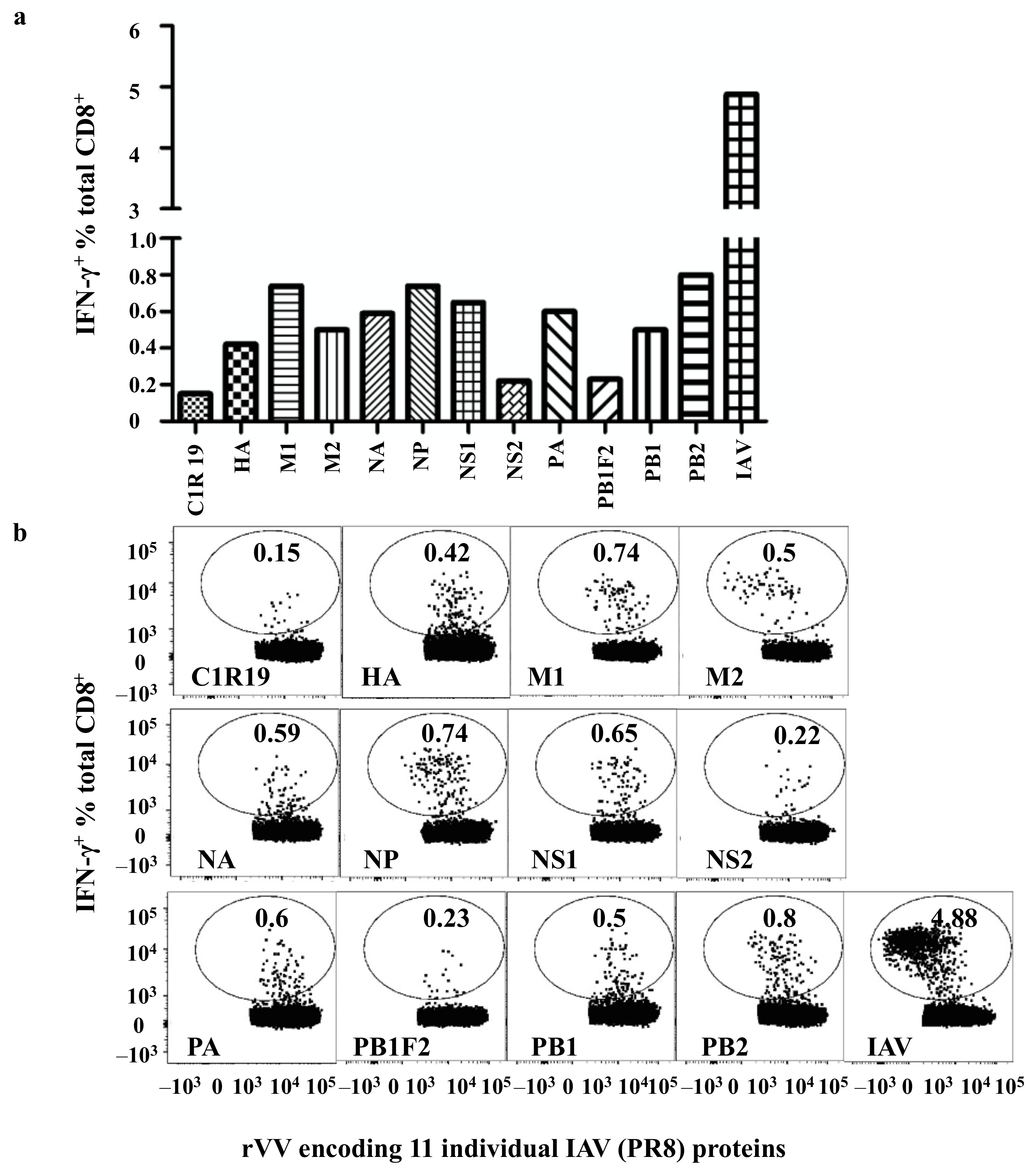
3.2. Broad-Based IAV-Specific CD8+ T Cell Responses to Multiple IAV Proteins
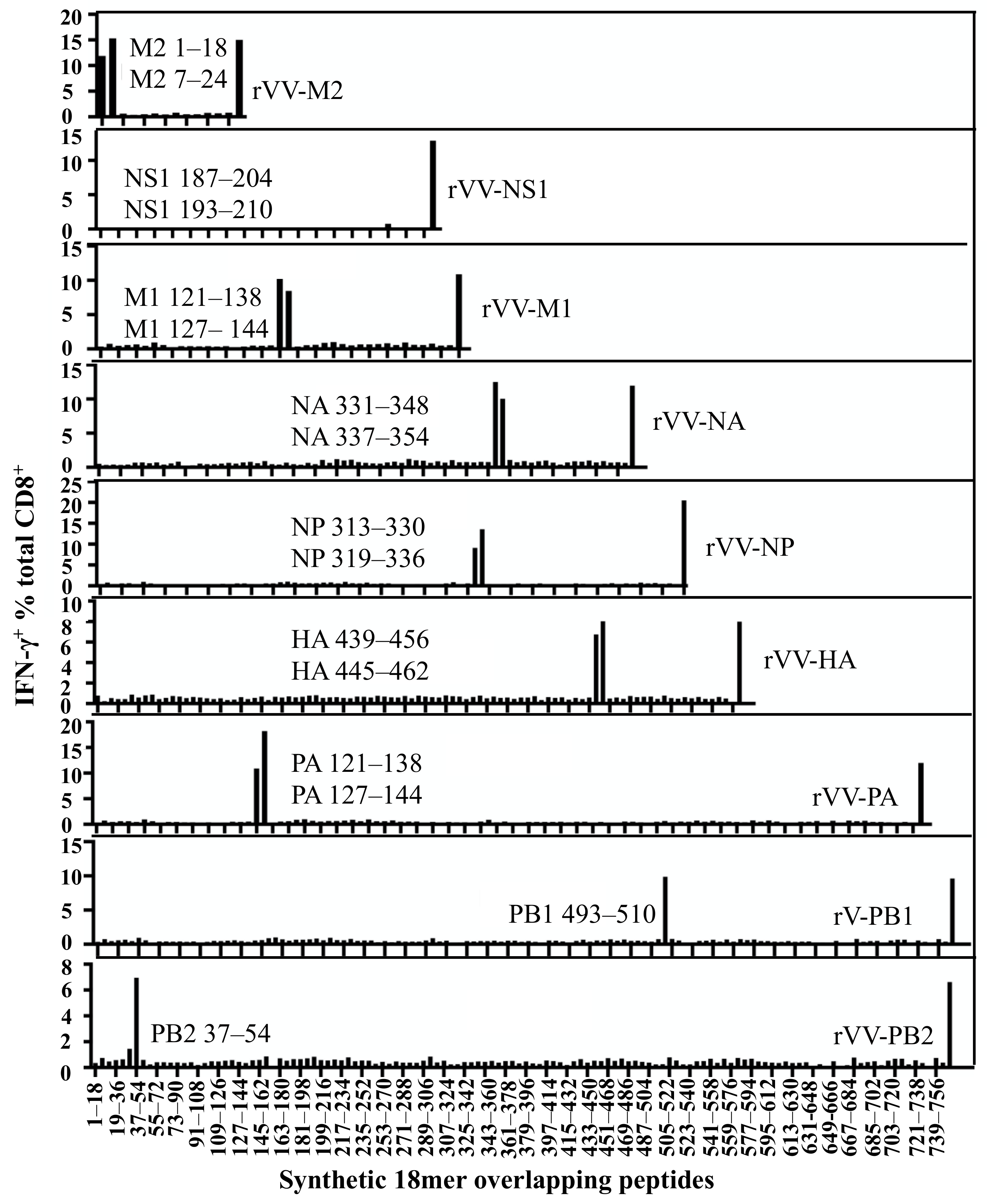
3.3. Characterising Antigenic Peptide Regions in Multiple IAV Proteins
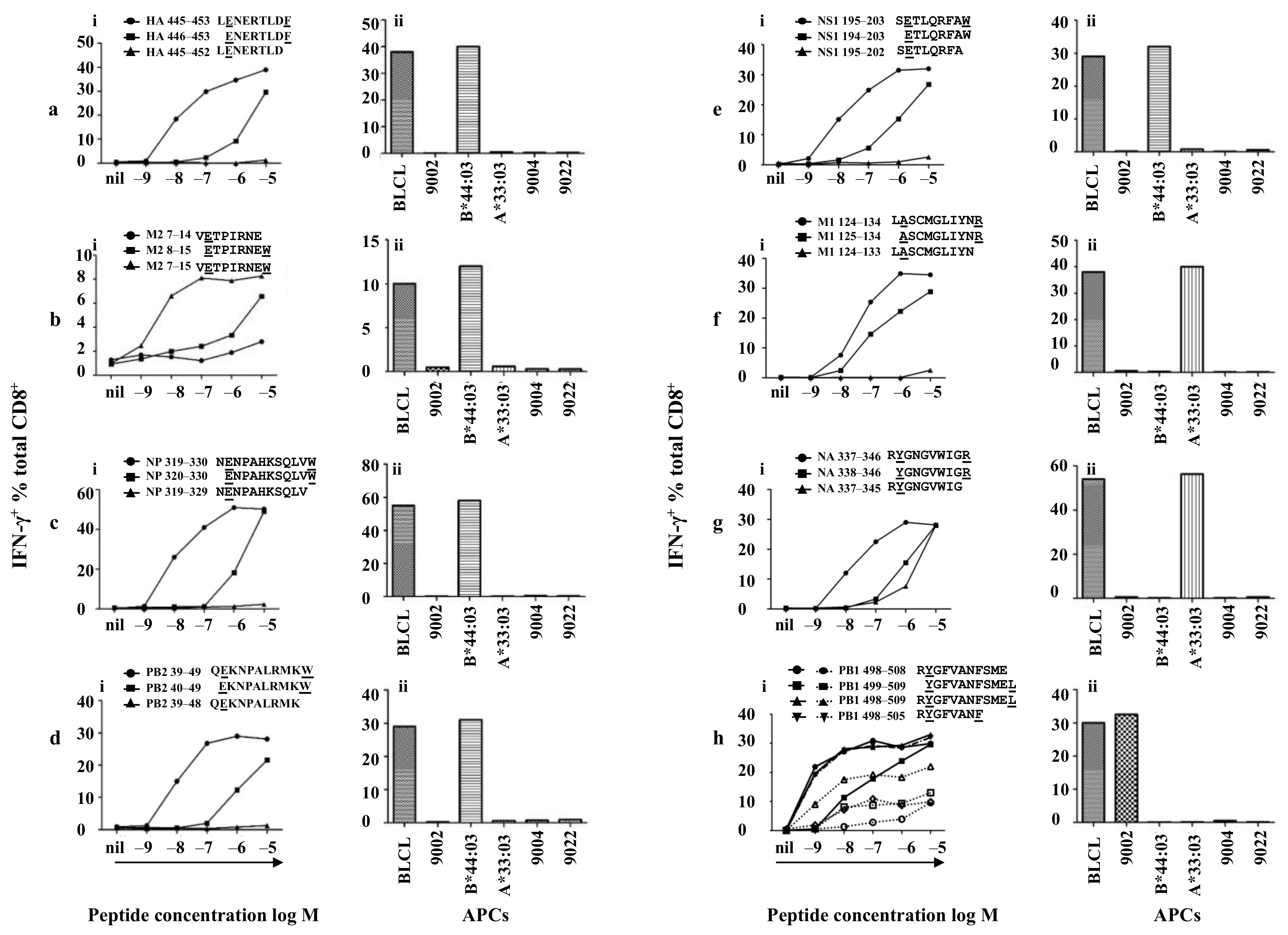
3.4. Minimal Epitopes of Differing Lengths Are Presented by Three HLA Alleles
3.5. Identifying the Core Sequences of the Epitopes Presented by HLA-B*44:03
3.6. Identifying the Core Sequences of the Epitopes Presented by HLA-A*33:03
3.7. Identifying the Core Sequences of the Epitopes Presented by HLA-A*24:02
3.8. Epitope Conservancy Among All the IAV Strains Circulated in Australia during 1971–2021 Period
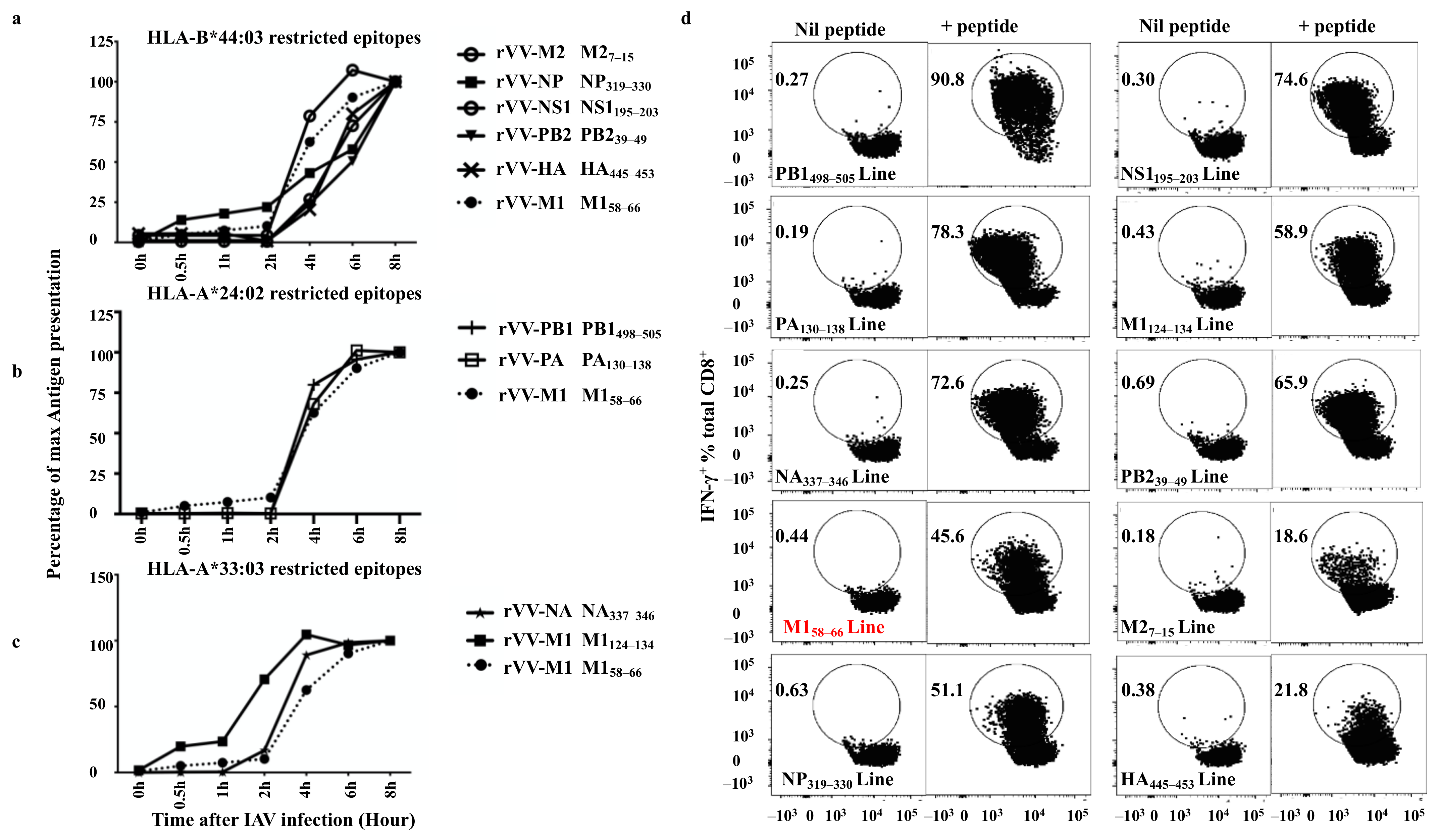
3.9. Quantitative Assessment of HLA-I Peptide Presentation to the Antigen-Specific CD8+ T Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmed, R.; Oldstone, M.B.; Palese, P. Protective immunity and susceptibility to infectious diseases: Lessons from the 1918 influenza pandemic. Nat. Immunol. 2007, 8, 1188–1193. [Google Scholar] [CrossRef]
- Grant, E.J.; Chen, L.; Quinones-Parra, S.; Pang, K.; Kedzierska, K.; Chen, W. T-cell immunity to influenza A viruses. Crit. Rev. Immunol. 2014, 34, 15–39. [Google Scholar] [CrossRef]
- Bender, B.S.; Croghan, T.; Zhang, L.; Small, P.A., Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J. Exp. Med. 1992, 175, 1143–1145. [Google Scholar] [CrossRef]
- Doherty, P.C.; Topham, D.J.; Tripp, R.A. Establishment and persistence of virus-specific CD4+ and CD8+ T cell memory. Immunol. Rev. 1996, 150, 23–44. [Google Scholar] [CrossRef]
- Wu, C.; Zanker, D.; Valkenburg, S.; Tan, B.; Kedzierska, K.; Zou, Q.M.; Doherty, P.C.; Chen, W. Systematic identification of immunodominant CD8+ T-cell responses to influenza A virus in HLA-A2 individuals. Proc. Natl. Acad. Sci. USA 2011, 108, 9178–9183. [Google Scholar] [CrossRef]
- Grant, E.; Wu, C.; Chan, K.F.; Eckle, S.; Bharadwaj, M.; Zou, Q.M.; Kedzierska, K.; Chen, W. Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T-cell responses. Immunol. Cell Biol. 2013, 91, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wan, Y.; Qiu, C.; Quinones-Parra, S.; Zhu, Z.; Loh, L.; Tian, D.; Ren, Y.; Hu, Y.; Zhang, X.; et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat. Commun. 2015, 6, 6833. [Google Scholar] [CrossRef]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef]
- Sercarz, E.E.; Lehmann, P.V.; Ametani, A.; Benichou, G.; Miller, A.; Moudgil, K. Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol. 1993, 11, 729–766. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Bennink, J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999, 17, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Anton, L.C.; Bennink, J.R.; Yewdell, J.W. Dissecting the multifactorial causes of immunodominance in class I-restricted T cell responses to viruses. Immunity 2000, 12, 83–93. [Google Scholar] [CrossRef]
- Belz, G.T.; Stevenson, P.G.; Doherty, P.C. Contemporary analysis of MHC-related immunodominance hierarchies in the CD8+ T cell response to influenza A viruses. J. Immunol. 2000, 165, 2404–2409. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Lewicki, H.; Borrow, P.; Hudrisier, D.; Gairin, J.E. Discriminated selection among viral peptides with the appropriate anchor residues: Implications for the size of the cytotoxic T-lymphocyte repertoire and control of viral infection. J. Virol. 1995, 69, 7423–7429. [Google Scholar] [CrossRef] [PubMed]
- Tscharke, D.C.; Karupiah, G.; Zhou, J.; Palmore, T.; Irvine, K.R.; Haeryfar, S.M.; Williams, S.; Sidney, J.; Sette, A.; Bennink, J.R.; et al. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J. Exp. Med. 2005, 201, 95–104. [Google Scholar] [CrossRef]
- Wallace, M.E.; Keating, R.; Heath, W.R.; Carbone, F.R. The cytotoxic T-cell response to herpes simplex virus type 1 infection of C57BL/6 mice is almost entirely directed against a single immunodominant determinant. J. Virol. 1999, 73, 7619–7626. [Google Scholar] [CrossRef] [PubMed]
- Boon, A.C.; de Mutsert, G.; Graus, Y.M.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. The magnitude and specificity of influenza A virus-specific cytotoxic T-lymphocyte responses in humans is related to HLA-A and -B phenotype. J. Virol. 2002, 76, 582–590. [Google Scholar] [CrossRef]
- Boon, A.C.; De Mutsert, G.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. Preferential HLA usage in the influenza virus-specific CTL response. J. Immunol. 2004, 172, 4435–4443. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Anthony, A.; Oveissi, S.; Huang, M.; Zanker, D.; Xiao, K.; Wu, C.; Zou, Q.; Chen, W. Broad-Based CD4+ T Cell Responses to Influenza A Virus in a Healthy Individual Who Lacks Typical Immunodominance Hierarchy. Front. Immunol. 2017, 8, 375. [Google Scholar] [CrossRef]
- Pang, K.C.; Wei, J.Q.; Chen, W. Dynamic quantification of MHC class I-peptide presentation to CD8+ T cells via intracellular cytokine staining. J. Immunol. Methods 2006, 311, 12–18. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, J.; Chen, W. FACS isolation of low percentage human antigen-specific CD8+ T cells based on activation-induced CD3 and CD8 downregulation. J. Immunol. Methods 2019, 472, 35–43. [Google Scholar] [CrossRef]
- Gotch, F.; Rothbard, J.; Howland, K.; Townsend, A.; McMichael, A. Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature 1987, 326, 881–882. [Google Scholar] [CrossRef]
- Burrows, S.R.; Silins, S.L.; Moss, D.J.; Khanna, R.; Misko, I.S.; Argaet, V.P. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J. Exp. Med. 1995, 182, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef]
- Rist, M.J.; Theodossis, A.; Croft, N.P.; Neller, M.A.; Welland, A.; Chen, Z.; Sullivan, L.C.; Burrows, J.M.; Miles, J.J.; Brennan, R.M.; et al. HLA peptide length preferences control CD8+ T cell responses. J. Immunol. 2013, 191, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Luescher, I.F.; Romero, P.; Kuznetsov, D.; Rimoldi, D.; Coulie, P.; Cerottini, J.C.; Jongeneel, C.V. HLA photoaffinity labeling reveals overlapping binding of homologous melanoma-associated gene peptides by HLA-A1, HLA-A29, and HLA-B44. J. Biol. Chem. 1996, 271, 12463–12471. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, M.; Matsuda, T.; Tomiyama, H.; Miwa, K. Analysis of three HLA-A*3303 binding peptide anchors using an HLA-A*3303 stabilization assay. Tissue Antigens 2000, 55, 296–302. [Google Scholar] [CrossRef]
- Kubo, R.T.; Sette, A.; Grey, H.M.; Appella, E.; Sakaguchi, K.; Zhu, N.Z.; Arnott, D.; Sherman, N.; Shabanowitz, J.; Michel, H.; et al. Definition of specific peptide motifs for four major HLA-A alleles. J. Immunol. 1994, 152, 3913–3924. [Google Scholar]
- Zanker, D.J.; Oveissi, S.; Tscharke, D.C.; Duan, M.; Wan, S.; Zhang, X.; Xiao, K.; Mifsud, N.A.; Gibbs, J.; Izzard, L.; et al. Influenza A Virus Infection Induces Viral and Cellular Defective Ribosomal Products Encoded by Alternative Reading Frames. J. Immunol. 2019, 202, 3370–3380. [Google Scholar] [CrossRef]
- Michaux, A.; Larrieu, P.; Stroobant, V.; Fonteneau, J.F.; Jotereau, F.; Van den Eynde, B.J.; Moreau-Aubry, A.; Vigneron, N. A spliced antigenic peptide comprising a single spliced amino acid is produced in the proteasome by reverse splicing of a longer peptide fragment followed by trimming. J. Immunol. 2014, 192, 1962–1971. [Google Scholar] [CrossRef]
- Chen, W.; Yewdell, J.W.; Levine, R.L.; Bennink, J.R. Modification of cysteine residues in vitro and in vivo affects the immunogenicity and antigenicity of major histocompatibility complex class I-restricted viral determinants. J. Exp. Med. 1999, 189, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, S.; Tan, S.; Yi, Y.; Wu, B.; Cao, B.; Zhu, F.; Wang, C.; Wang, H.; Qi, J.; et al. Cross-allele cytotoxic T lymphocyte responses against 2009 pandemic H1N1 influenza A virus among HLA-A24 and HLA-A3 supertype-positive individuals. J. Virol. 2012, 86, 13281–13294. [Google Scholar] [CrossRef][Green Version]
- Falo, L.D., Jr.; Colarusso, L.J.; Benacerraf, B.; Rock, K.L. Serum proteases alter the antigenicity of peptides presented by class I major histocompatibility complex molecules. Proc. Natl. Acad. Sci. USA 1992, 89, 834–850. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Bilsel, P.; del Guercio, M.F.; Marinkovic-Petrovic, A.; Southwood, S.; Stewart, S.; Ishioka, G.; Kotturi, M.F.; Botten, J.; Sidney, J.; et al. Identification of broad binding class I HLA supertype epitopes to provide universal coverage of influenza A virus. Hum. Immunol. 2010, 71, 468–474. [Google Scholar] [CrossRef]
- Tenzer, S.; Wee, E.; Burgevin, A.; Stewart-Jones, G.; Friis, L.; Lamberth, K.; Chang, C.H.; Harndahl, M.; Weimershaus, M.; Gerstoft, J.; et al. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat. Immunol. 2009, 10, 636–646. [Google Scholar] [CrossRef]
- Akram, A.; Inman, R.D. Immunodominance: A pivotal principle in host response to viral infections. Clin. Immunol. 2012, 143, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pang, K.; Masterman, K.A.; Kennedy, G.; Basta, S.; Dimopoulos, N.; Hornung, F.; Smyth, M.; Bennink, J.R.; Yewdell, J.W. Reversal in the immunodominance hierarchy in secondary CD8+ T cell responses to influenza A virus: Roles for cross-presentation and lysis-independent immunodomination. J. Immunol. 2004, 173, 5021–5027. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Bennink, J.R. Brefeldin A specifically inhibits presentation of protein antigens to cytotoxic T lymphocytes. Science 1989, 244, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Quinones-Parra, S.; Grant, E.; Loh, L.; Nguyen, T.H.; Campbell, K.A.; Tong, S.Y.; Miller, A.; Doherty, P.C.; Vijaykrishna, D.; Rossjohn, J.; et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc. Natl. Acad. Sci. USA 2014, 111, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Hertz, T.; Oshansky, C.M.; Roddam, P.L.; DeVincenzo, J.P.; Caniza, M.A.; Jojic, N.; Mallal, S.; Phillips, E.; James, I.; Halloran, M.E.; et al. HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection. Proc. Natl. Acad. Sci. USA 2013, 110, 13492–13497. [Google Scholar] [CrossRef]
- Clemens, E.B.; Grant, E.J.; Wang, Z.; Gras, S.; Tipping, P.; Rossjohn, J.; Miller, A.; Tong, S.Y.; Kedzierska, K. Towards identification of immune and genetic correlates of severe influenza disease in Indigenous Australians. Immunol. Cell Biol. 2016, 94, 367–377. [Google Scholar] [CrossRef]
- Burrows, S.R.; Rodda, S.J.; Suhrbier, A.; Geysen, H.M.; Moss, D.J. The specificity of recognition of a cytotoxic T lymphocyte epitope. Eur. J. Immunol. 1992, 22, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Flint, S.M.; Davis, J.S.; Su, J.Y.; Oliver-Landry, E.P.; Rogers, B.A.; Goldstein, A.; Thomas, J.H.; Parameswaran, U.; Bigham, C.; Freeman, K.; et al. Disproportionate impact of pandemic (H1N1) 2009 influenza on Indigenous people in the Top End of Australia’s Northern Territory. Med. J. Aust. 2010, 192, 617–622. [Google Scholar] [CrossRef]
- Kelly, H.; Mercer, G.; Cheng, A. Quantifying the risk of pandemic influenza in pregnancy and indigenous people in Australia in 2009. Eurosurveillance 2009, 14, 19441. [Google Scholar]
- Trauer, J.M.; Laurie, K.L.; McDonnell, J.; Kelso, A.; Markey, P.G. Differential effects of pandemic (H1N1) 2009 on remote and indigenous groups, Northern Territory, Australia, 2009. Emerg. Infect. Dis. 2011, 17, 1615–1623. [Google Scholar] [PubMed]
- Thomas, P.G.; Brown, S.A.; Keating, R.; Yue, W.; Morris, M.Y.; So, J.; Webby, R.J.; Doherty, P.C. Hidden epitopes emerge in secondary influenza virus-specific CD8+ T cell responses. J. Immunol. 2007, 178, 3091–3098. [Google Scholar] [CrossRef]
- Kim, P.S.; Lee, P.P.; Levy, D. A theory of immunodominance and adaptive regulation. Bull. Math. Biol. 2011, 73, 1645–1665. [Google Scholar] [CrossRef]
- Kedl, R.M.; Rees, W.A.; Hildeman, D.A.; Schaefer, B.; Mitchell, T.; Kappler, J.; Marrack, P. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 2000, 192, 1105–1113. [Google Scholar] [CrossRef]
- Wallace, M.E.; Bryden, M.; Cose, S.C.; Coles, R.M.; Schumacher, T.N.; Brooks, A.; Carbone, F.R. Junctional biases in the naive TCR repertoire control the CTL response to an immunodominant determinant of HSV-1. Immunity 2000, 12, 547–556. [Google Scholar] [CrossRef]
- Day, E.B.; Charlton, K.L.; La Gruta, N.L.; Doherty, P.C.; Turner, S.J. Effect of MHC class I diversification on influenza epitope-specific CD8+ T cell precursor frequency and subsequent effector function. J. Immunol. 2011, 186, 6319–6328. [Google Scholar] [CrossRef]
- Neller, M.A.; Ladell, K.; McLaren, J.E.; Matthews, K.K.; Gostick, E.; Pentier, J.M.; Dolton, G.; Schauenburg, A.J.; Koning, D.; Fontaine Costa, A.I.; et al. Naive CD8+ T-cell precursors display structured TCR repertoires and composite antigen-driven selection dynamics. Immunol. Cell Biol. 2015, 93, 625–633. [Google Scholar] [CrossRef]
- Akram, A.; Inman, R.D. Co-expression of HLA-B7 and HLA-B27 alleles is associated with B7-restricted immunodominant responses following influenza infection. Eur. J. Immunol. 2013, 43, 3254–3267. [Google Scholar] [CrossRef] [PubMed]
- Bihl, F.; Frahm, N.; Di Giammarino, L.; Sidney, J.; John, M.; Yusim, K.; Woodberry, T.; Sango, K.; Hewitt, H.S.; Henry, L.; et al. Impact of HLA-B alleles, epitope binding affinity, functional avidity, and viral coinfection on the immunodominance of virus-specific CTL responses. J. Immunol. 2006, 176, 4094–4101. [Google Scholar] [CrossRef] [PubMed]
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory inflation: Continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 2003, 170, 2022–2029. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APCs | HLA-A | HLA-B | HLA-Cw |
|---|---|---|---|
| auto-BLCL | 24:02, 33:03 | 44:03, 46:01 | 01:02, 07:01 |
| 9002 | 24:02 | 14:02 | 02:02, 08:02 |
| C1R-B*44:03 | 44:03, residual 35:03 | 04:01 | |
| C1R-A*33:03 | 33:03 | residual 35:03 | 04:01 |
| 9004 | 02:01 | 27:05 | 01:02 |
| 9022 | 01:01 | 08:01 | 07:01 |
 |
| H1N1 | H3N2 | ||
|---|---|---|---|
| A/Cairns North/INS597/2010 | A/Australia/18/2009 | A/Perth/1055/2014 | A/Sydney/852M/2002 |
| A/Sydney/DD3-02/2009 | A/Australia/NHRC0001/2003 | A/Perth/113/2015 | A/Sydney/329Q/2003 |
| A/Victoria/2005/2009 | A/Australia/NHRC0001/2005 | A/Perth/153/2012 | A/Sydney/419J/2004 |
| A/Brisbane/59/2007 | A/Brisbane/9/2006 | A/Perth/16/2009 | A/Sydney/348N/2006 |
| A/New South Wales/18/1999(H1N1) | A/Brisbane/1/2012 | A/Perth/164/2013 | A/Sydney/257U/2009 |
| A/South Australia/45/2000 | A/Brisbane/1/2013 | A/Perth/22/2016 | A/Sydney/DD2-01/2010 |
| A/Western Australia/18/2001 | A/Brisbane/1/2017 | A/Queensland/1/2000 | A/Tasmania/60/2012 |
| A/Brisbane/193/2004 | A/Brisbane/10/2007 | A/Queensland/11/2001 | A/Tasmania/11/2014 |
| A/South Australia/55/2005 | A/Brisbane/100/2014 | A/Queensland/21/2002 | A/Tasmania/1005/2015 |
| A/Western Australia/77/2005 | A/Brisbane/1000/2015 | A/Queensland/31/2003 | A/Tasmania/1001/2016 |
| A/Victoria/500/2006 | A/Brisbane/1004/2016 | A/Queensland/41/2004 | A/Townsville/1004/2012 |
| A/Brisbane/297/2006 | A/Brisbane/11/2010 | A/Queensland/51/2005 | A/Townsville/10/2015 |
| A/Perth/1/2007 | A/Canberra/1001/2012 | A/South Australia/1/2012 | A/Townsville/1003/2016 |
| A/Victoria/501/2007 | A/Canberra/1001/2016 | A/South Australia/1/2015 | A/Victoria/75/1995 |
| A/South Australia/423/2007 | A/Canberra/113/2013 | A/South Australia/1002/2014 | A/Victoria/210/2009 |
| A/Sydney/581/2007 | A/Canberra/12/2015 | A/South Australia/1007/2016 | A/Victoria/361/2011 |
| A/South Australia/2001/2009 | A/Canberra/2/2014 | A/South Australia/15/1994 | A/Victoria/1002/2012 |
| A/Brisbane/17/2009 | A/Darwin/1000/2015 | A/South Australia/15/2000 | A/Victoria/504/2013 |
| A/Australia/1/2009 | A/Darwin/1001/2012 | A/South Australia/18/2005 | A/Victoria/1012/2014 |
| A/swine/VIC/09-02767-01/2009 | A/Darwin/1002/2016 | A/South Australia/2/2013 | A/Victoria/1000/2015 |
| A/swine/QLD/09-02865-07/2009 | A/Darwin/4/2005 | A/Sydney/223G/2005 | A/Victoria/109/2016 |
| A/Sydney/DD3-37/2010 | A/New South Wales/13/1999 | A/Sydney/0017/2007 | A/Western Australia/1/2000 |
| A/North Fitzroy/INS463/2010 | A/New South Wales/27/2000 | A/Sydney/1001/2012 | A/Western Australia/13/2001 |
| A/Melbourne/INS467/2010 | A/Newcastle/1/2013 | A/Sydney/1014/2013 | A/Western Australia/23/2002 |
| A/Westmead/INS524/2010 | A/Newcastle/1000/2016 | A/Sydney/1004/2014 | A/Western Australia/37/2003 |
| A/Sydney/DD3-58/2011 | A/Newcastle/1001/2015 | A/Sydney/10/2015 | A/Western Australia/51/2004 |
| A/Westmead/INS3_659/2011 | A/Newcastle/1016/2014 | A/Sydney/101/2016 | A/Western Australia/65/2005 |
| A/Darlinghurst/INS3_645/2011 | A/PERTH/1/2003 | A/Sydney/5/1997 | |
| A/Melbourne/INS3_670/2011 | A/Perth/10/2010 | A/Sydney/405A/2001 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Xu, R.; Triffon, C.; Mifsud, N.; Chen, W. Broad-Based Influenza-Specific CD8+ T Cell Response without the Typical Immunodominance Hierarchy and Its Potential Implication. Viruses 2021, 13, 1080. https://doi.org/10.3390/v13061080
Huang M, Xu R, Triffon C, Mifsud N, Chen W. Broad-Based Influenza-Specific CD8+ T Cell Response without the Typical Immunodominance Hierarchy and Its Potential Implication. Viruses. 2021; 13(6):1080. https://doi.org/10.3390/v13061080
Chicago/Turabian StyleHuang, Miaojuan, Rong Xu, Cristina Triffon, Nicole Mifsud, and Weisan Chen. 2021. "Broad-Based Influenza-Specific CD8+ T Cell Response without the Typical Immunodominance Hierarchy and Its Potential Implication" Viruses 13, no. 6: 1080. https://doi.org/10.3390/v13061080
APA StyleHuang, M., Xu, R., Triffon, C., Mifsud, N., & Chen, W. (2021). Broad-Based Influenza-Specific CD8+ T Cell Response without the Typical Immunodominance Hierarchy and Its Potential Implication. Viruses, 13(6), 1080. https://doi.org/10.3390/v13061080