
Molecular Characterization of Highly Pathogenic Avian Influenza Viruses H5N6 Detected in Denmark in 2018–2019

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Virus Detection, Subtyping and Antibody Detection
2.3. Sequencing and Consensus Sequence Generation
2.4. Data Selection
2.5. Bayesian Phylogenetic Analysis
2.6. Molecular Clock Analysis
3. Results
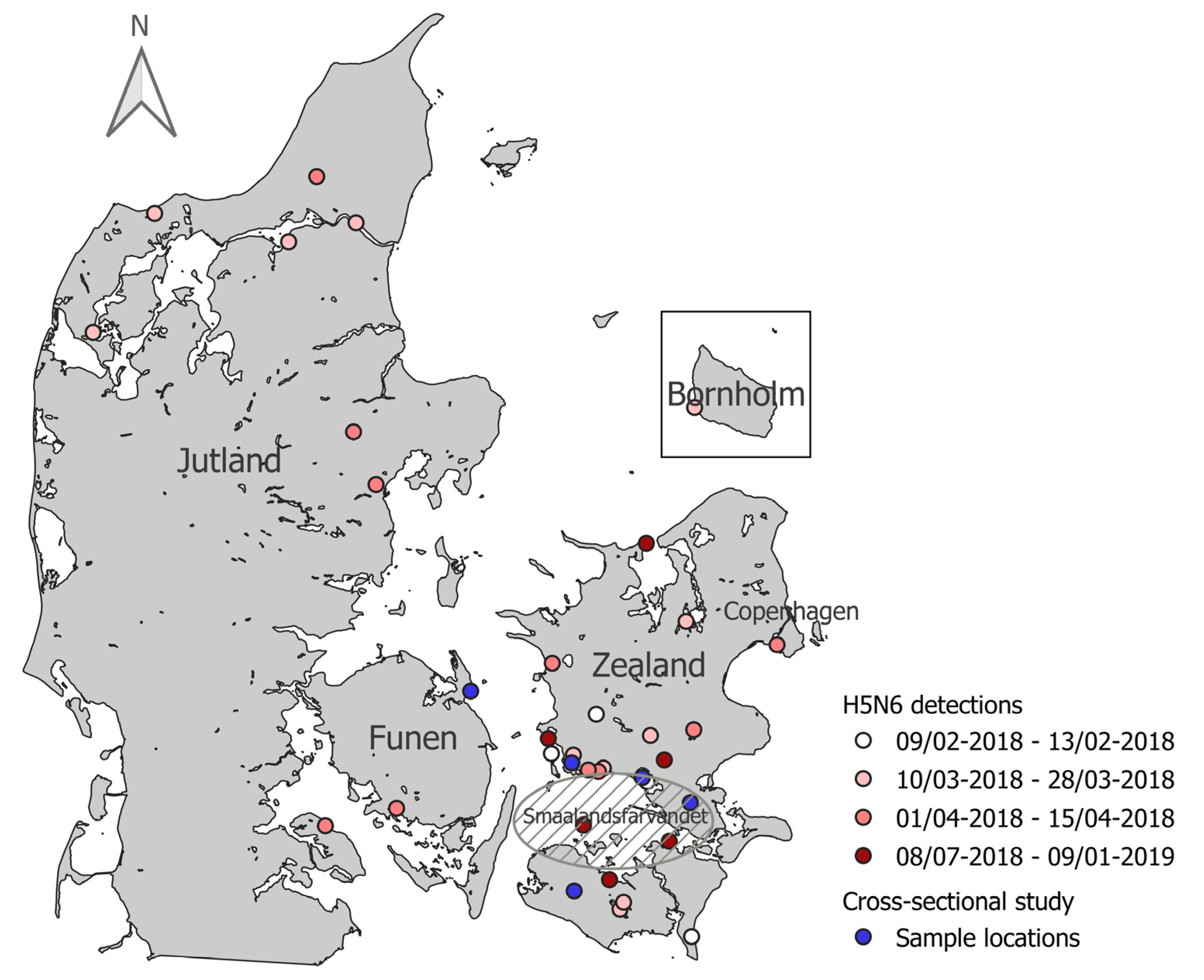
3.1. AIV Wild Bird Active Surveillance
3.2. AIV Wild Bird Passive Surveillance
3.3. HPAI Virus Prevalence in Clinically Healthy Wild Pheasants
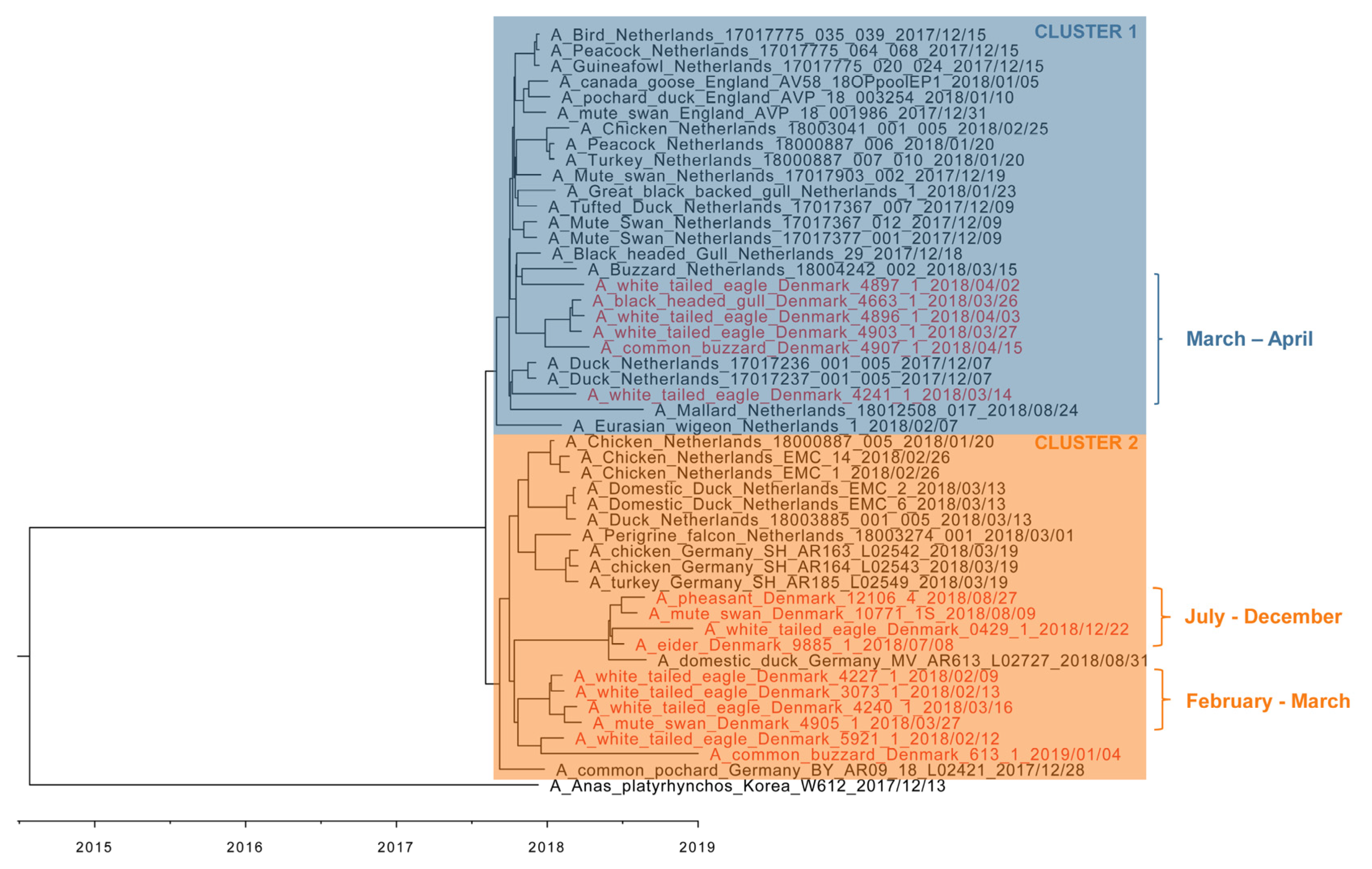
3.4. Phylogenetic Analyses
3.5. Molecular Dating
3.6. Zoonotic Potential
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef]
- World Health Organization (WHO); World Organisation for Animal Health/Food (OIE); Agriculture Organisation (FAO); H5N1 Evolution Working Group. Revised and updated nomenclature for highly pathogenic avian influenza A (H5N1) viruses. Influenza Other Respi. Viruses 2014, 8, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Bertran, K.; Kwon, J.H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Poen, M.J.; Venkatesh, D.; Bestebroer, T.M.; Vuong, O.; Scheuer, R.D.; Oude Munnink, B.B.; de Meulder, D.; Richard, M.; Kuiken, T.; Koopmans, M.P.G.; et al. Co-circulation of genetically distinct highly pathogenic avian influenza A clade 2.3.4.4 (H5N6) viruses in wild waterfowl and poultry in Europe and East Asia, 2017–18. Virus Evol. 2019, 5, 1–12. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Kuiken, T.; Niqueux, E.; Staubach, C.; Terregino, C.; Guajardo, I.M.; et al. Avian influenza overview November 2019–February 2020. EFSA J. 2020, 18, e06096. [Google Scholar] [CrossRef] [PubMed]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Kuiken, T.; Niqueux, E.; Staubach, C.; Terregino, C.; Guajardo, I.M.; et al. Avian influenza overview February–May 2020. EFSA J. 2020, 18, e06194. [Google Scholar] [CrossRef] [PubMed]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Kuiken, T.; Niqueux, É.; Staubach, C.; Terregino, C.; Muñoz Guajardo, I.; et al. Avian influenza overview May–August 2020. EFSA J. 2020, 18, e06270. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Avian influenza overview August–December 2020. EFSA J. 2020, 18, e06379. [Google Scholar] [CrossRef] [PubMed]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Avian influenza overview December 2020–February 2021. EFSA J. 2021, 19, e06497. [Google Scholar] [CrossRef]
- The Global Consortium for H5N8 and Related Influenza Viruses Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–218. [CrossRef]
- Verhagen, J.H.; Herfst, S.; Fouchier, R.A.M. How a virus travels the world. Science 2015, 347, 616–617. [Google Scholar] [CrossRef] [PubMed]
- Mine, J.; Uchida, Y.; Sharshov, K.; Sobolev, I.; Shestopalov, A.; Saito, T. Phylogeographic evidence for the inter- and intracontinental dissemination of avian influenza viruses via migration flyways. PLoS ONE 2019, 14, e0218506. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Heutink, R.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Multiple reassorted viruses as cause of highly pathogenic avian influenza A(H5N8) virus epidemic, the Netherlands, 2016. Emerg. Infect. Dis. 2017, 23, 1966–1973. [Google Scholar] [CrossRef]
- Globig, A.; Staubach, C.; Sauter-Louis, C.; Dietze, K.; Homeier-Bachmann, T.; Probst, C.; Gethmann, J.; Depner, K.R.; Grund, C.; Harder, T.C.; et al. Highly pathogenic Avian influenza H5N8 clade 2.3.4.4b in Germany in 2016/2017. Front. Vet. Sci. 2018, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Napp, S.; Majó, N.; Sánchez-Gónzalez, R.; Vergara-Alert, J. Emergence and spread of highly pathogenic avian influenza A(H5N8) in Europe in 2016-2017. Transbound. Emerg. Dis. 2018, 65, 1217–1226. [Google Scholar] [CrossRef]
- World Organization for Animal Health. Immediate Notification and Follow-Up Reports of Highly Influenza (Types H5 and H7). Available online: https://www.oie.int/en/animal-health-in-the-world/update-on-avian-influenza/ (accessed on 25 August 2020).
- Beerens, N.; Koch, G.; Heutink, R.; Harders, F.; Vries, D.P.E.; Ho, C.; Bossers, A.; Elbers, A. Novel highly pathogenic avian influenza A(H5N6) viruses in the Netherlands, December 2017. Emerg. Infect. Dis. 2018, 24, 770–773. [Google Scholar] [CrossRef]
- Miljø- og Fødevareministeriet. Bekendtgørelse om Udsætning af Vildt, Jagtmåder og Jagtredskaber; BEK nr 1652 af 19/12/2017; Miljø- og Fødevareministeriet, 2017. [Google Scholar]
- Humberd, J.; Boyd, K.; Webster, R.G. Emergence of Influenza A Virus Variants after Prolonged Shedding from Pheasants. J. Virol. 2007, 81, 4044–4051. [Google Scholar] [CrossRef]
- Humberd, J.; Guan, Y.; Webster, R.G. Comparison of the Replication of Influenza A Viruses in Chinese Ring-Necked Pheasants and Chukar Partridges. J. Virol. 2006, 80, 2151–2161. [Google Scholar] [CrossRef]
- World Health Organization. World Health Organization. Avian Influenza Weekly Update Number 788. Human Infection with Avian Influenza A (H5) viruses. 2021. Available online: http://www.who.int.
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory on Avian Influenza); Brown, I.; Kuiken, T.; Mulatti, P.; Smietanka, K.; Staubach, C.; Stroud, D.; Therkildsen, O.R.; et al. Avian influenza overview September–November 2017. EFSA J. 2017, 15. [Google Scholar] [CrossRef]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef]
- Slomka, M.J.; To, T.L.; Tong, H.H.; Coward, V.J.; Hanna, A.; Shell, W.; Pavlidis, T.; Densham, A.L.E.; Kargiolakis, G.; Arnold, M.E.; et al. Challenges for accurate and prompt molecular diagnosis of clades of highly pathogenic avian influenza H5N1 viruses emerging in Vietnam. Avian Pathol. 2012, 41, 177–193. [Google Scholar] [CrossRef]
- Slomka, A.M.J.; Pavlidis, T.; Banks, J.; Shell, W.; Mcnally, A.; Essen, S.; Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; et al. Validated H5 Eurasian Real-Time Reverse Transcriptase-Polymerase Chain Reaction and Its Application in H5N1 Outbreaks in 2005-2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef]
- Slomka, M.J.; Pavlidis, T.; Coward, V.J.; Voermans, J.; Koch, G.; Hanna, A.; Banks, J.; Brown, I.H. Validated RealTime reverse transcriptase PCR methods for the diagnosis and pathotyping of Eurasian H7 avian influenza viruses. Influenza Other Respi. Viruses 2009, 3, 151–164. [Google Scholar] [CrossRef]
- Slomka, M.J.; Coward, V.J.; Banks, J.; Löndt, B.Z.; Brown, I.H.; Voermans, J.; Koch, G.; Handberg, K.J.; Jørgensen, P.H.; Cherbonnel-Pansart, M.; et al. Identification of sensitive and specific avian influenza polymerase chain reaction methods through blind ring trials organized in the European Union. Avian Dis. 2007, 51, 227–234. [Google Scholar] [CrossRef]
- Lee, H.K.; Tang, J.W.T.; Kong, D.H.L.; Koay, E.S.C. Simplified Large-Scale Sanger Genome Sequencing for Influenza A/H3N2 Virus. PLoS ONE 2013, 8, e64785. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Nissen, J.N.; Krog, J.S.; Breum, S.Ø.; Trebbien, R.; Larsen, L.E.; Hjulsager, C.K. Novel Clade 2.3.4.4b Highly Pathogenic Avian Influenza A(H5N8 and H5N5) Viruses in Denmark, 2020. Viruses 2021, 13, 886. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree Version 1.4.4. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/.
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- QGIS Development Team 2021. QGIS Geographic Information System. Open Source Geospatial Foundation Project. Version 3.14.1. Available online: http://qgis.osgeo.org.
- Pohlmann, A.; Hoffmann, D.; Grund, C.; Koethe, S.; Hüssy, D.; Meier, S.M.; King, J.; Schinköthe, J.; Ulrich, R.; Harder, T.; et al. Genetic characterization and zoonotic potential of highly pathogenic avian influenza virus A(H5N6/H5N5), Germany, 2017–2018. Emerg. Infect. Dis. 2019, 25, 1973–1976. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Heutink, R.; Pritz-Verschuren, S.; Germeraad, E.A.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Genetic relationship between poultry and wild bird viruses during the highly pathogenic avian influenza H5N6 epidemic in the Netherlands, 2017–2018. Transbound. Emerg. Dis. 2019, 66, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- EU. Annual Report on Surveillance for Avian Influenza in Poultry and Wild Birds in Member States of the European Union in 2017; EU: Brussels, Belgium, 2018; pp. 1–165. [Google Scholar]
- Hjulsager, C.K.; Breum, S.Ø.; Trebbien, R.; Handberg, K.J.; Therkildsen, O.R.; Madsen, J.J.; Thorup, K.; Baroch, J.A.; DeLiberto, T.J.; Larsen, L.E.; et al. Surveillance for Avian Influenza Viruses in Wild Birds in Denmark and Greenland, 2007–10. Avian Dis. 2012, 56, 992–998. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Node | tMRCA | 95% HPD Interval | Posterior Probability | Origin | |
|---|---|---|---|---|---|
| PB2 | 1 | April 2017 | January 2017–August 2017 | 1.0000 | European LPAI |
| 2 | May 2010 | November 2009–November 2010 | 0.5341 | ||
| 3 | February 2010 | August 2009–August 2010 | 0.9228 | ||
| HA | 1 | July 2017 | May 2017–September 2017 | 0.5786 | H5N8 HPAI 2016–2017 |
| 2 | June 2017 | March 2017–August 2017 | 1.0000 | ||
| 3 | September 2016 | July 2016–November 2016 | 0.311 | ||
| NA | 1 | January 2017 | October 2016–May 2017 | 0.7741 | H5N6 HPAI 2017 |
| 2 | December 2016 | August 2016–April 2017 | 1.0000 | European LPAI | |
| 3 | July 2015 | December 2014–January 2016 | 1.0000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Krog, J.S.; Ryt-Hansen, P.; Pedersen, A.G.; Kvisgaard, L.K.; Holm, E.; Nielsen, P.D.; Hammer, A.S.; Madsen, J.J.; Thorup, K.; et al. Molecular Characterization of Highly Pathogenic Avian Influenza Viruses H5N6 Detected in Denmark in 2018–2019. Viruses 2021, 13, 1052. https://doi.org/10.3390/v13061052
Liang Y, Krog JS, Ryt-Hansen P, Pedersen AG, Kvisgaard LK, Holm E, Nielsen PD, Hammer AS, Madsen JJ, Thorup K, et al. Molecular Characterization of Highly Pathogenic Avian Influenza Viruses H5N6 Detected in Denmark in 2018–2019. Viruses. 2021; 13(6):1052. https://doi.org/10.3390/v13061052
Chicago/Turabian StyleLiang, Yuan, Jesper Schak Krog, Pia Ryt-Hansen, Anders Gorm Pedersen, Lise Kirstine Kvisgaard, Elisabeth Holm, Pernille Dahl Nielsen, Anne Sofie Hammer, Jesper Johannes Madsen, Kasper Thorup, and et al. 2021. "Molecular Characterization of Highly Pathogenic Avian Influenza Viruses H5N6 Detected in Denmark in 2018–2019" Viruses 13, no. 6: 1052. https://doi.org/10.3390/v13061052
APA StyleLiang, Y., Krog, J. S., Ryt-Hansen, P., Pedersen, A. G., Kvisgaard, L. K., Holm, E., Nielsen, P. D., Hammer, A. S., Madsen, J. J., Thorup, K., Larsen, L. E., & Hjulsager, C. K. (2021). Molecular Characterization of Highly Pathogenic Avian Influenza Viruses H5N6 Detected in Denmark in 2018–2019. Viruses, 13(6), 1052. https://doi.org/10.3390/v13061052