
Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition


Abstract
1. Introduction
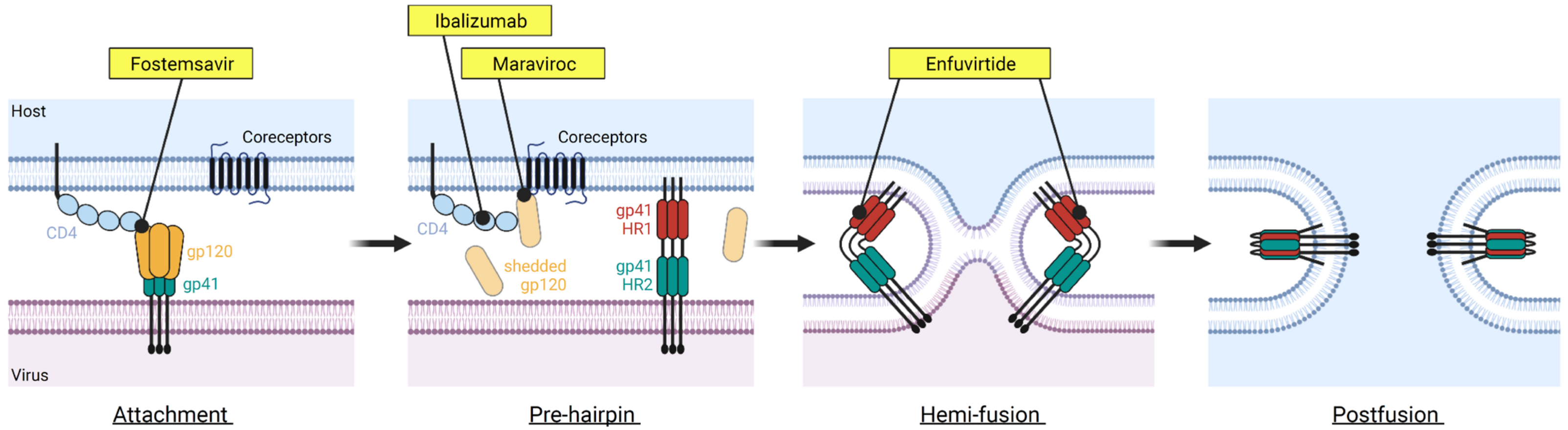
2. HIV-1 Entry into Host Cells
FDA-Approved Drugs Targeting HIV-1 Entry
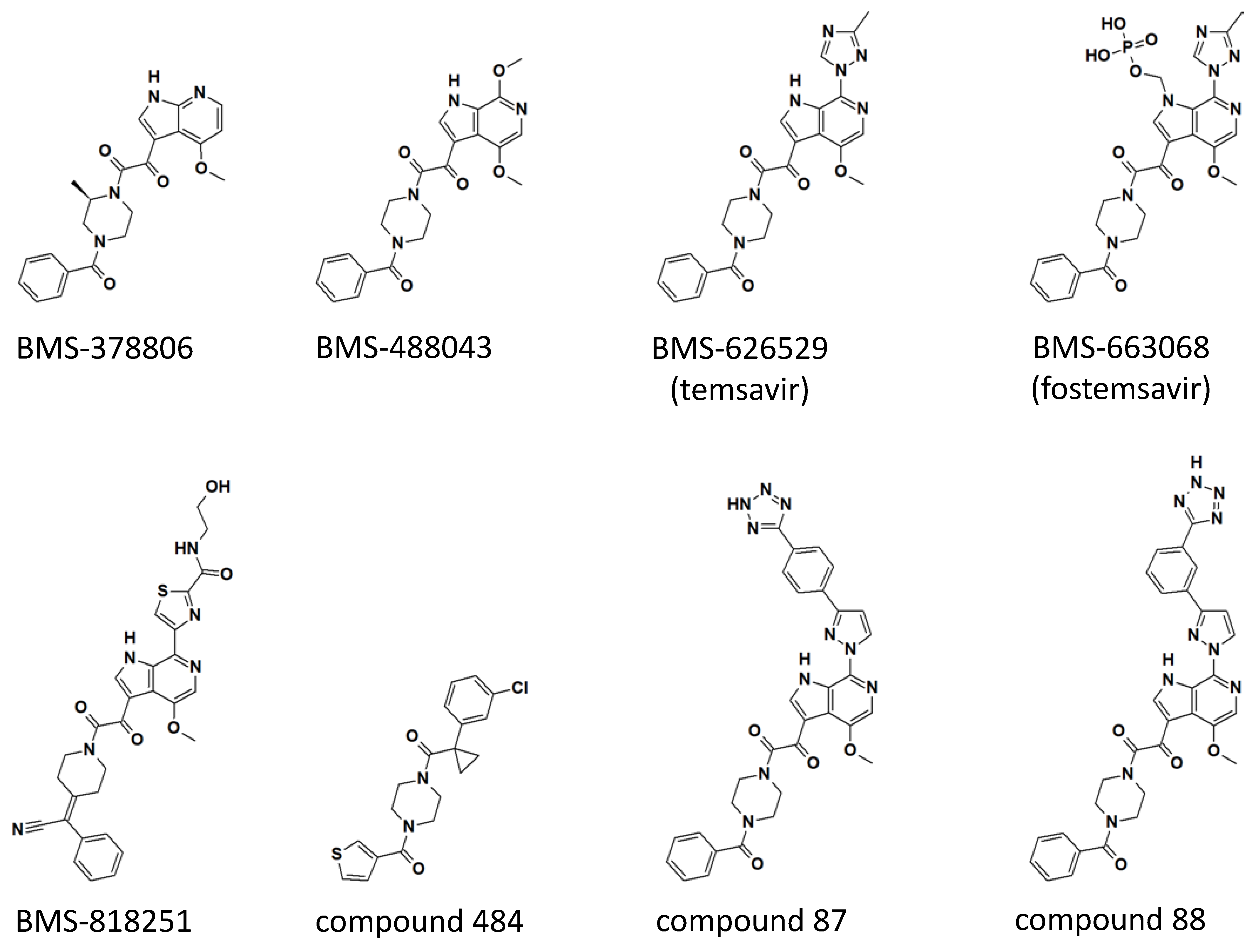
3. Discovery, Clinical Trials, and FDA Approval of Fostemsavir
Clinical Development of Indole Glyoxamide Inhibitors
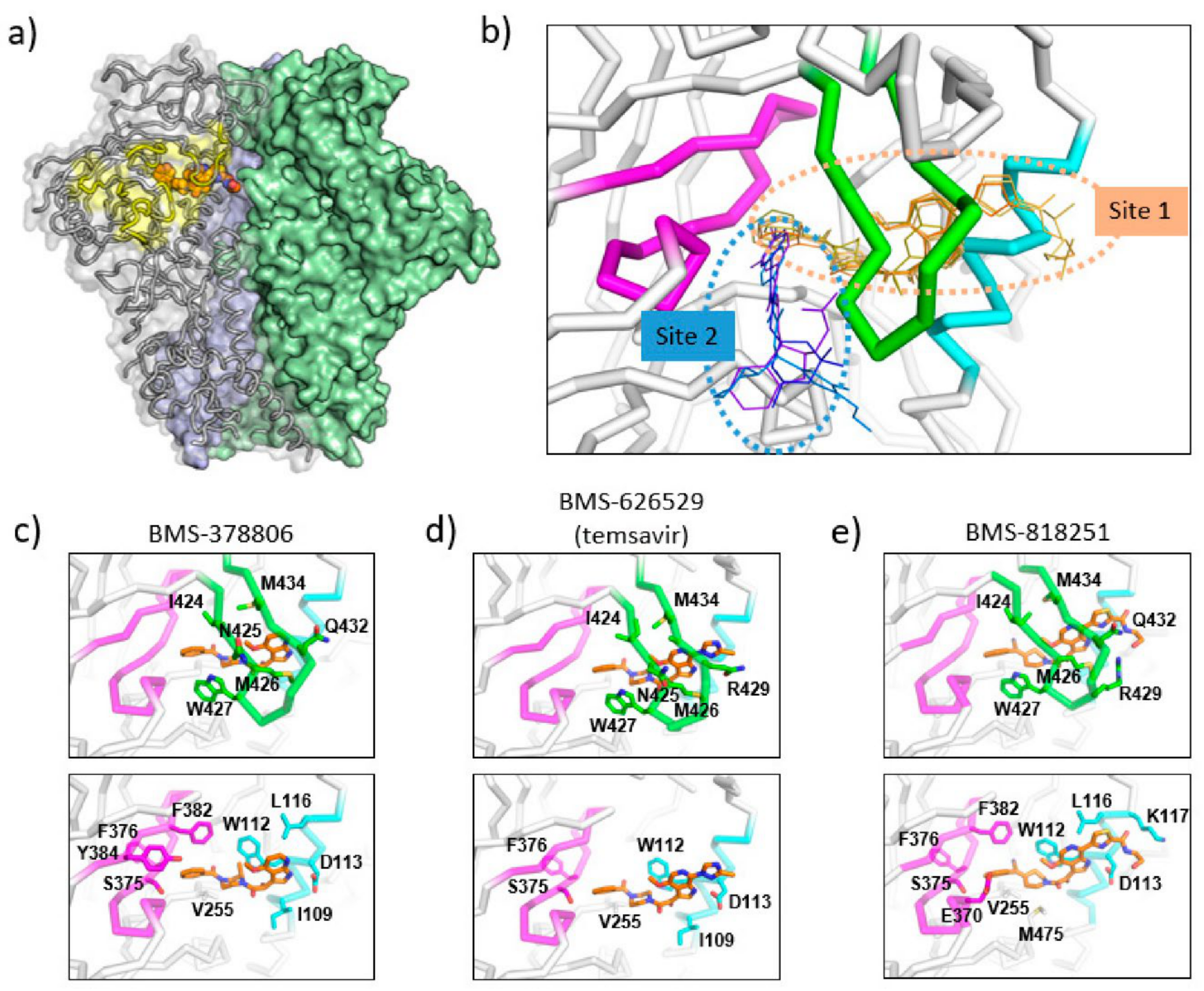
4. Mode of Action and Structural Basis of Inhibition for Temsavir and Analogues
5. Next Generation Inhibitors: Potent Temsavir Analogues
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naif, H.M. Pathogenesis of HIV infection. Infect. Dis. Rep. 2013, 5, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef]
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr. HIV Res. 2018, 15, 411–421. [Google Scholar] [CrossRef]
- De Bethune, M.P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antivir. Res. 2010, 85, 75–90. [Google Scholar] [CrossRef]
- Messiaen, P.; Wensing, A.M.J.; Fun, A.; Nijhuis, M.; Brusselaers, N.; Vandekerckhove, L. Clinical Use of HIV Integrase Inhibitors: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e52562. [Google Scholar] [CrossRef]
- Scarsi, K.K.; Havens, J.P.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 2020, 80, 1649–1676. [Google Scholar] [CrossRef]
- Emu, B.; Fessel, J.; Schrader, S.; Kumar, P.; Richmond, G.; Win, S.; Weinheimer, S.; Marsolais, C.; Lewis, S. Phase 3 Study of Ibalizumab for Multidrug-Resistant HIV-1. N. Engl. J. Med. 2018, 379, 645–654. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Woollard, S.M. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015, 9, 5447–5468. [Google Scholar] [CrossRef]
- Zhang, D.; Li, W.; Jiang, S. Peptide fusion inhibitors targeting the HIV-1 gp41: A patent review (2009–2014). Expert Opin. Ther. Pat. 2014, 25, 159–173. [Google Scholar] [CrossRef]
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 drug resistance and resistance testing. Infect. Genet. Evol. 2016, 46, 292–307. [Google Scholar] [CrossRef]
- Teeraananchai, S.; Kerr, S.J.; Amin, J.; Ruxrungtham, K.; Law, M.G. Life expectancy of HIV-positive people after starting combination antiretroviral therapy: A meta-analysis. HIV Med. 2017, 18, 256–266. [Google Scholar] [CrossRef]
- Feng, Q.; Zhou, A.; Zou, H.; Ingle, S.; May, M.T.; Cai, W.; Cheng, C.-Y.; Yang, Z.; Tang, J. Quadruple versus triple combination antiretroviral therapies for treatment naive people with HIV: Systematic review and meta-analysis of randomised controlled trials. BMJ 2019, 366, l4179. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 Envelope Glycoprotein Biosynthesis, Trafficking, and Incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef]
- Pezeshkian, N.; Groves, N.S.; Van Engelenburg, S.B. Single-molecule imaging of HIV-1 envelope glycoprotein dynamics and Gag lattice association exposes determinants responsible for virus incorporation. Proc. Natl. Acad. Sci. USA 2019, 116, 25269–25277. [Google Scholar] [CrossRef]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef]
- Ward, A.B.; A Wilson, I. The HIV-1 envelope glycoprotein structure: Nailing down a moving target. Immunol. Rev. 2017, 275, 21–32. [Google Scholar] [CrossRef]
- Piai, A.; Fu, Q.; Cai, Y.; Ghantous, F.; Xiao, T.; Shaik, M.; Peng, H.; Rits-Volloch, S.; Chen, W.; Seaman, M.S.; et al. Structural basis of transmembrane coupling of the HIV-1 envelope glycoprotein. Nat. Commun. 2020, 11, 2317. [Google Scholar] [CrossRef]
- Clapham, P.R.; McKnight, Á. HIV-1 receptors and cell tropism. Br. Med. Bull. 2001, 58, 43–59. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, H.; Liu, A.Z.; Gristick, H.B.; Bjorkman, P.J. Asymmetric opening of HIV-1 Env bound to CD4 and a coreceptor-mimicking antibody. Nat. Struct. Mol. Biol. 2019, 26, 1167–1175. [Google Scholar] [CrossRef]
- Liu, Q.; Acharya, P.; Dolan, M.A.; Zhang, P.; Guzzo, C.; Lu, J.; Kwon, A.; Gururani, D.; Miao, H.; Bylund, T.; et al. Quaternary contact in the initial interaction of CD4 with the HIV-1 envelope trimer. Nat. Struct. Mol. Biol. 2017, 24, 370–378. [Google Scholar] [CrossRef]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef]
- Klasse, P.J. The molecular basis of HIV entry. Cell. Microbiol. 2012, 14, 1183–1192. [Google Scholar] [CrossRef]
- Lu, L.; Yu, F.; Cai, L.; Debnath, A.K.; Jiang, S. Development of Small-molecule HIV Entry Inhibitors Specifically Targeting gp120 or gp41. Curr. Top. Med. Chem. 2015, 16, 1074–1090. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Zhang, W.; et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Joly, V.; Jidar, K.; Tatay, M.; Yeni, P. Enfuvirtide: From basic investigations to current clinical use. Expert Opin. Pharmacother. 2010, 11, 2701–2713. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.M.; Seaman, M.S.; Rits-Volloch, S.; Hong, X.; Kao, C.-Y.; Ho, D.D.; Chen, B. Crystal Structure of HIV-1 Primary Receptor CD4 in Complex with a Potent Antiviral Antibody. Structure 2010, 18, 1632–1641. [Google Scholar] [CrossRef]
- Lin, P.-F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.-F.; Wang, H.-G.H.; Rose, R.; Yamanaka, G.; et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. USA 2003, 100, 11013–11018. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-Benzoyl-1-[(4-methoxy-1H- pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2-(R)-methylpiperazine (BMS-378806): A Novel HIV-1 Attachment Inhibitor That Interferes with CD4-gp120 Interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ueda, Y.; Zhang, Z.; Yin, Z.; Matiskella, J.; Pearce, B.C.; Yang, Z.; Zheng, M.; Parker, D.D.; Yamanaka, G.A.; et al. Discovery of the Human Immunodeficiency Virus Type 1 (HIV-1) Attachment Inhibitor Temsavir and Its Phosphonooxymethyl Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 6308–6327. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Ho, H.-T.; Dicker, I.; Fan, L.; Zhou, N.; Friborg, J.; Wang, T.; McAuliffe, B.V.; Wang, H.-G.H.; Rose, R.E.; et al. Biochemical and Genetic Characterizations of a Novel Human Immunodeficiency Virus Type 1 Inhibitor That Blocks gp120-CD4 Interactions. J. Virol. 2003, 77, 10528–10536. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.L.; Cilliers, T.; Morris, L. Predicted genotypic resistance to the novel entry inhibitor, BMS-378806, among HIV-1 isolates of subtypes A to G. AIDS 2004, 18, 2327–2330. [Google Scholar] [CrossRef]
- Xue, Y.-J.; Yan, J.-H.; Arnold, M.; Grasela, D.; Unger, S. Quantitative determination of BMS-378806 in human plasma and urine by high-performance liquid chromatography/tandem mass spectrometry. J. Sep. Sci. 2007, 30, 1267–1275. [Google Scholar] [CrossRef]
- Kadow, J.F.; Ueda, Y.; Meanwell, N.A.; Connolly, T.P.; Wang, T.; Chen, C.-P.; Yeung, K.-S.; Zhu, J.; Bender, J.A.; Yang, Z.; et al. Inhibitors of Human Immunodeficiency Virus Type 1 (HIV-1) Attachment 6. Preclinical and Human Pharmacokinetic Profiling of BMS-663749, a Phosphonooxymethyl Prodrug of the HIV-1 Attachment Inhibitor 2-(4-Benzoyl-1-piperazinyl)-1-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxoethanone (BMS-488043). J. Med. Chem. 2012, 55, 2048–2056. [Google Scholar] [CrossRef]
- Hanna, G.J.; Lalezari, J.; Hellinger, J.A.; Wohl, D.A.; Nettles, R.; Persson, A.; Krystal, M.; Lin, P.; Colonno, R.; Grasela, D.M. Antiviral Activity, Pharmacokinetics, and Safety of BMS-488043, a Novel Oral Small-Molecule HIV-1 Attachment Inhibitor, in HIV-1-Infected Subjects. Antimicrob. Agents Chemother. 2011, 55, 722–728. [Google Scholar] [CrossRef]
- Zhou, N.; Nowicka-Sans, B.; Zhang, S.; Fan, L.; Fang, J.; Fang, H.; Gong, Y.-F.; Eggers, B.; Langley, D.R.; Wang, T.; et al. In Vivo Patterns of Resistance to the HIV Attachment Inhibitor BMS-488043. Antimicrob. Agents Chemother. 2010, 55, 729–737. [Google Scholar] [CrossRef]
- Ueda, Y.; Matiskella, J.D.; Golik, J.; Connolly, T.P.; Hudyma, T.W.; Venkatesh, S.; Dali, M.; Kang, S.-H.; Barbour, N.; Tejwani, R.; et al. Phosphonooxymethyl prodrugs of the broad spectrum antifungal azole, ravuconazole: Synthesis and biological properties. Bioorg. Med. Chem. Lett. 2003, 13, 3669–3672. [Google Scholar] [CrossRef]
- Liu, T.; Huang, B.; Zhan, P.; De Clercq, E.; Liu, X. Discovery of small molecular inhibitors targeting HIV-1 gp120–CD4 interaction drived from BMS-378806. Eur. J. Med. Chem. 2014, 86, 481–490. [Google Scholar] [CrossRef]
- Swidorski, J.J.; Liu, Z.; Yin, Z.; Wang, T.; Carini, D.J.; Rahematpura, S.; Zheng, M.; Johnson, K.; Zhang, S.; Lin, P.-F.; et al. Inhibitors of HIV-1 attachment: The discovery and structure–activity relationships of tetrahydroisoquinolines as replacements for the piperazine benzamide in the 3-glyoxylyl 6-azaindole pharmacophore. Bioorg. Med. Chem. Lett. 2016, 26, 160–167. [Google Scholar] [CrossRef]
- Schader, S.M.; Colby-Germinario, S.P.; Quashie, P.K.; Oliveira, M.; Ibanescu, R.-I.; Moisi, D.; Mespléde, T.; Wainberg, M.A. HIV gp120 H375 Is Unique to HIV-1 Subtype CRF01_AE and Confers Strong Resistance to the Entry Inhibitor BMS-599793, a Candidate Microbicide Drug. Antimicrob. Agents Chemother. 2012, 56, 4257–4267. [Google Scholar] [CrossRef]
- Nowicka-Sans, B.; Gong, Y.-F.; McAuliffe, B.; Dicker, I.; Ho, H.-T.; Zhou, N.; Eggers, B.; Lin, P.-F.; Ray, N.; Wind-Rotolo, M.; et al. In Vitro Antiviral Characteristics of HIV-1 Attachment Inhibitor BMS-626529, the Active Component of the Prodrug BMS-663068. Antimicrob. Agents Chemother. 2012, 56, 3498–3507. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef]
- Nettles, R.E.; Schürmann, D.; Zhu, L.; Stonier, M.; Huang, S.-P.; Chang, I.; Chien, C.; Krystal, M.; Wind-Rotolo, M.; Ray, N.; et al. Pharmacodynamics, Safety, and Pharmacokinetics of BMS-663068, an Oral HIV-1 Attachment Inhibitor in HIV-1-Infected Subjects. J. Infect. Dis. 2012, 206, 1002–1011. [Google Scholar] [CrossRef]
- Lataillade, M.; Zhou, N.; Joshi, S.R.; Lee, S.; Stock, D.A.; Hanna, G.J.; Krystal, M. Viral Drug Resistance through 48 Weeks, in a Phase 2b, Randomized, Controlled Trial of the HIV-1 Attachment Inhibitor Prodrug, Fostemsavir. JAIDS J. Acquir. Immune Defic. Syndr. 2018, 77, 299–307. [Google Scholar] [CrossRef]
- Kozal, M.; Aberg, J.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.-M.; Grinsztejn, B.; Diaz, R.; Castagna, A.; Kumar, P.; et al. Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2020, 382, 1232–1243. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, N.; Sun, Y.; Ray, N.; Lataillade, M.; Hanna, G.J.; Krystal, M. Activity of the HIV-1 Attachment Inhibitor BMS-626529, the Active Component of the Prodrug BMS-663068, against CD4-Independent Viruses and HIV-1 Envelopes Resistant to Other Entry Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 4172–4180. [Google Scholar] [CrossRef]
- Si, Z.; Madani, N.; Cox, J.M.; Chruma, J.J.; Klein, J.C.; Schön, A.; Phan, N.; Wang, L.; Biorn, A.C.; Cocklin, S.; et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 5036–5041. [Google Scholar] [CrossRef]
- Kong, R.; Tan, J.J.; Ma, X.H.; Zu Chen, W.; Wang, C.X. Prediction of the binding mode between BMS-378806 and HIV-1 gp120 by docking and molecular dynamics simulation. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2006, 1764, 766–772. [Google Scholar] [CrossRef]
- Langley, D.R.; Kimura, S.R.; Sivaprakasam, P.; Zhou, N.; Dicker, I.; McAuliffe, B.; Wang, T.; Kadow, J.F.; Meanwell, N.A.; Krystal, M. Homology models of the HIV-1 attachment inhibitor BMS-626529 bound to gp120 suggest a unique mechanism of action. Proteins Struct. Funct. Bioinform. 2015, 83, 331–350. [Google Scholar] [CrossRef]
- Pancera, M.; Lai, Y.-T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schön, A.; Bailer, R.T.; Chuang, G.-Y.; Geng, H.; et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115–1122. [Google Scholar] [CrossRef]
- Lai, Y.-T.; Wang, T.; O’Dell, S.; Louder, M.K.; Schön, A.; Cheung, C.S.F.; Chuang, G.-Y.; Druz, A.; Lin, B.; McKee, K.; et al. Lattice engineering enables definition of molecular features allowing for potent small-molecule inhibition of HIV-1 entry. Nat. Commun. 2019, 10, 47. [Google Scholar] [CrossRef]
- Herschhorn, A.; Gu, C.; Moraca, F.; Ma, X.; Farrell, M.; Smith, A.B., III; Pancera, M.; Kwong, P.D.; Schon, A.; Freire, E.; et al. The beta20-beta21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat. Commun. 2017, 8, 1049. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.D.; Finzi, A.; Wu, X.; Dogo-Isonagie, C.; Lee, L.K.; Moore, L.R.; Schmidt, S.D.; Stuckey, J.; Yang, Y.; Zhou, T.; et al. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. USA 2012, 109, 5663–5668. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.D.; LaLonde, J.M.; Yang, Y.; Elban, M.A.; Sugawara, A.; Courter, J.R.; Jones, D.M.; Smith, A.B.; Debnath, A.K.; Kwong, P.D. Crystal Structures of HIV-1 gp120 Envelope Glycoprotein in Complex with NBD Analogues That Target the CD4-Binding Site. PLoS ONE 2014, 9, e85940. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, J.M.; Le-Khac, M.; Jones, D.M.; Courter, J.R.; Park, J.; Schön, A.; Princiotto, A.M.; Wu, X.; Mascola, J.R.; Freire, E.; et al. Structure-Based Design and Synthesis of an HIV-1 Entry Inhibitor Exploiting X-ray and Thermodynamic Characterization. ACS Med. Chem. Lett. 2013, 4, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Crowell, T.A.; Colby, D.J.; Pinyakorn, S.; Sacdalan, C.; Pagliuzza, A.; Intasan, J.; Benjapornpong, K.; Tangnaree, K.; Chomchey, N.; Kroon, E.; et al. Safety and efficacy of VRC01 broadly neutralising antibodies in adults with acutely treated HIV (RV397): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet HIV 2019, 6, e297–e306. [Google Scholar] [CrossRef]
- Riddler, S.A.; Zheng, L.; Durand, C.M.; Ritz, J.; Koup, R.A.; Ledgerwood, J.; Bailer, R.T.; Koletar, S.L.; Eron, J.J.; Keefer, M.C.; et al. Randomized Clinical Trial to Assess the Impact of the Broadly Neutralizing HIV-1 Monoclonal Antibody VRC01 on HIV-1 Persistence in Individuals on Effective ART. Open Forum Infect. Dis. 2018, 5, 242. [Google Scholar] [CrossRef]
- Wu, X. HIV Broadly Neutralizing Antibodies: VRC01 and Beyond. Adv. Exp. Med. Biol. 2018, 1075, 53–72. [Google Scholar] [CrossRef]
- Julg, B.; Pegu, A.; Abbink, P.; Liu, J.; Brinkman, A.; Molloy, K.; Mojta, S.; Chandrashekar, A.; Callow, K.; Wang, K.; et al. Virological Control by the CD4-Binding Site Antibody N6 in Simian-Human Immunodeficiency Virus-Infected Rhesus Monkeys. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Scheid, J.F.; Horwitz, J.A.; Bar-On, Y.; Kreider, E.F.; Lu, C.-L.; Lorenzi, J.C.C.; Feldmann, A.; Braunschweig, M.; Nogueira, L.; Oliveira, T.; et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 2016, 535, 556–560. [Google Scholar] [CrossRef]
- Cohen, Y.Z.; Butler, A.L.; Millard, K.; Witmer-Pack, M.; Levin, R.; Unson-O’Brien, C.; Patel, R.; Shimeliovich, I.; Lorenzi, J.C.C.; Horowitz, J.; et al. Safety, pharmacokinetics, and immunogenicity of the combination of the broadly neutralizing anti-HIV-1 antibodies 3BNC117 and 10-1074 in healthy adults: A randomized, phase 1 study. PLoS ONE 2019, 14, e0219142. [Google Scholar] [CrossRef]
- Caskey, M.; Klein, F.; Lorenzi, J.C.C.; Seaman, M.S.; West, A.P.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2015, 522, 487–491. [Google Scholar] [CrossRef]
- Fellinger, C.H.; Gardner, M.R.; Weber, J.A.; Alfant, B.; Zhou, A.S.; Farzan, M. eCD4-Ig Limits HIV-1 Escape More Effectively Than CD4-Ig or a Broadly Neutralizing Antibody. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Gardner, M.R.; Fellinger, C.H.; Kattenhorn, L.M.; Davis-Gardner, M.E.; Weber, J.A.; Alfant, B.; Zhou, A.S.; Prasad, N.R.; Kondur, H.R.; Newton, W.A.; et al. AAV-delivered ecd4-Ig protects rhesus macaques from high-dose sivmac239 challenges. Sci. Transl. Med. 2019, 11, eaau5409. [Google Scholar] [CrossRef]
- Gardner, M.R.; Farzan, M. Engineering antibody-like inhibitors to prevent and treat HIV-1 infection. Curr. Opin. HIV AIDS 2017, 12, 294–301. [Google Scholar] [CrossRef]
- Hoffmann, M.A.G.; Bar-On, Y.; Yang, Z.; Gristick, H.B.; Gnanapragasam, P.N.P.; Vielmetter, J.; Nussenzweig, M.C.; Bjorkman, P.J. Nanoparticles presenting clusters of CD4 expose a universal vulnerability of HIV-1 by mimicking target cells. Proc. Natl. Acad. Sci. USA 2020, 117, 18719–18728. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Class | Generic Name (Other Names and Acronyms) | Brand Name | |
|---|---|---|---|
| NRTIs | abacavir (abacavir sulfate, ABC) | Ziagen | |
| emtricitabine (FTC) | Emtriva | ||
| lamivudine (3TC) | Epivir | ||
| tenofovir disoproxil fumarate (tenofovir DF, TDF) | Viread | ||
| zidovudine (azidothymidine, AZT, ZDV) | Retrovir | ||
| NNRTIs | doravirine (DOR) | Pifeltro | |
| efavirenz (EFV) | Sustiva | ||
| etravirine (ETR) | Intelence | ||
| nevirapine (extended release nevirapine, NVP) | Viramune/XR | ||
| rilpivirine (rilpivirine hydrochloride, RPV) | Edurant | ||
| Protease Inhibitors | atazanavir (atazanavir sulfate, ATV) | Reyataz | |
| darunavir (darunavir ethanolate, DRV) | Prezista | ||
| fosamprenavir (fosamprenavir calcium, FOS-APV, FPV) | Lexiva | ||
| ritonavir (RTV) | Norvir | ||
| saquinavir (saquinavir mesylate, SQV) | Invirase | ||
| tipranavir (TPV) | Aptivus | ||
| Integrase Inhibitors | cabotegravir (cabotegravir sodium, CAB) | Vocabria | |
| dolutegravir (dolutegravir sodium, DTG) | Tivicay | ||
| raltegravir (raltegravir potassium, RAL) | Isentress | ||
| Entry Inhibitors | Fusion Inhibitors | enfuvirtide (T-20) | Fuzeon |
| CCR5 Antagonists | maraviroc (MVC) | Selzentry | |
| Post-attachment Inhibitors | ibalizumab-uiyk (Hu5A8, IBA, Ibalizumab, TMB-355, TNX-355) | Trogarzo | |
| Attachment Inhibitors | fostemsavir (fostemsavir tromethamine, FTR) | Rukobia | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, Y.-T. Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition. Viruses 2021, 13, 843. https://doi.org/10.3390/v13050843
Lai Y-T. Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition. Viruses. 2021; 13(5):843. https://doi.org/10.3390/v13050843
Chicago/Turabian StyleLai, Yen-Ting. 2021. "Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition" Viruses 13, no. 5: 843. https://doi.org/10.3390/v13050843
APA StyleLai, Y.-T. (2021). Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition. Viruses, 13(5), 843. https://doi.org/10.3390/v13050843