
Identification of the SHREK Family of Proteins as Broad-Spectrum Host Antiviral Factors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Plasmid Transfection and Virus Production
2.3. Virus Infectivity Assays
2.4. HIV Env Incorporation Assay
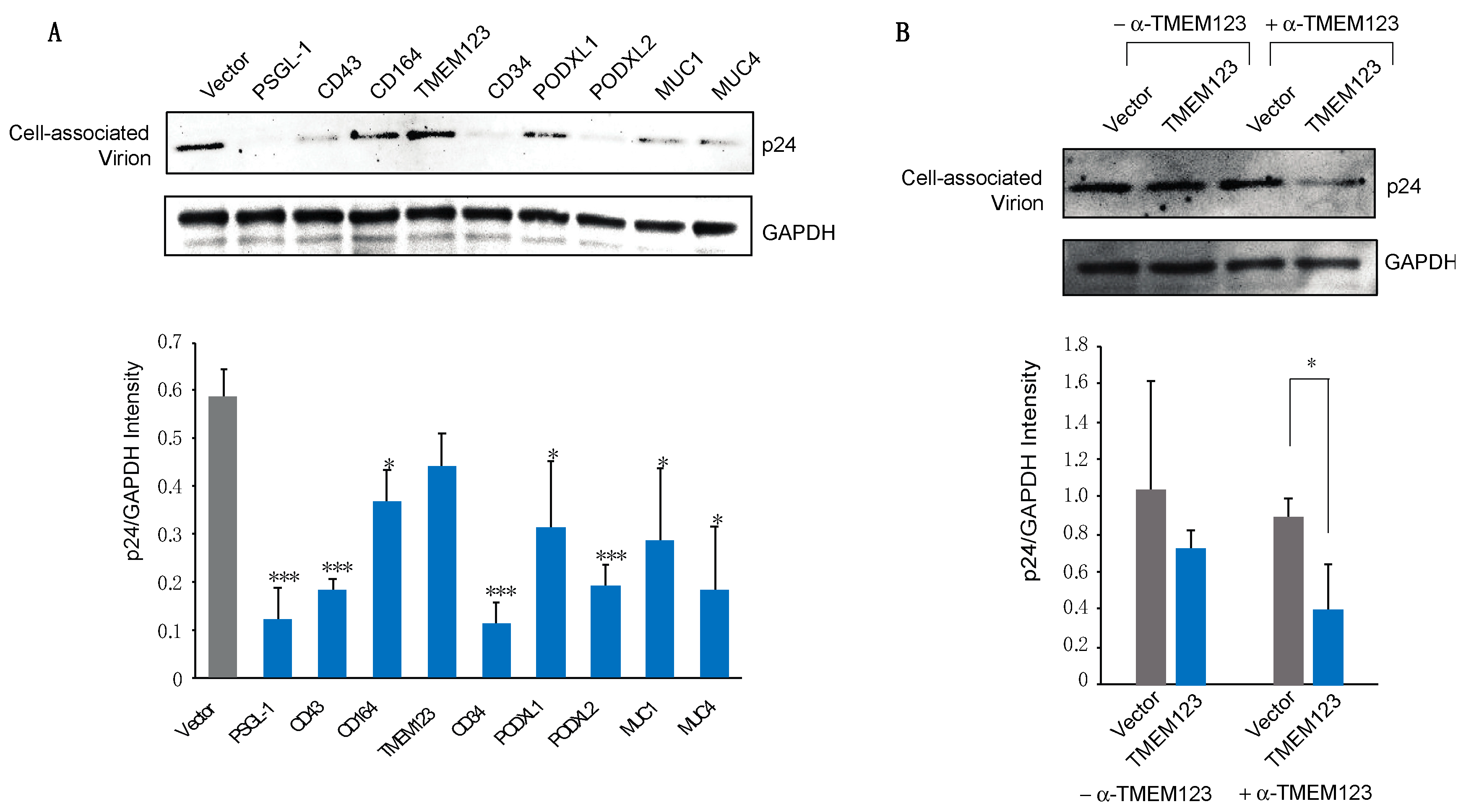
2.5. Detection of SHREK Proteins in HIV-1 Particles
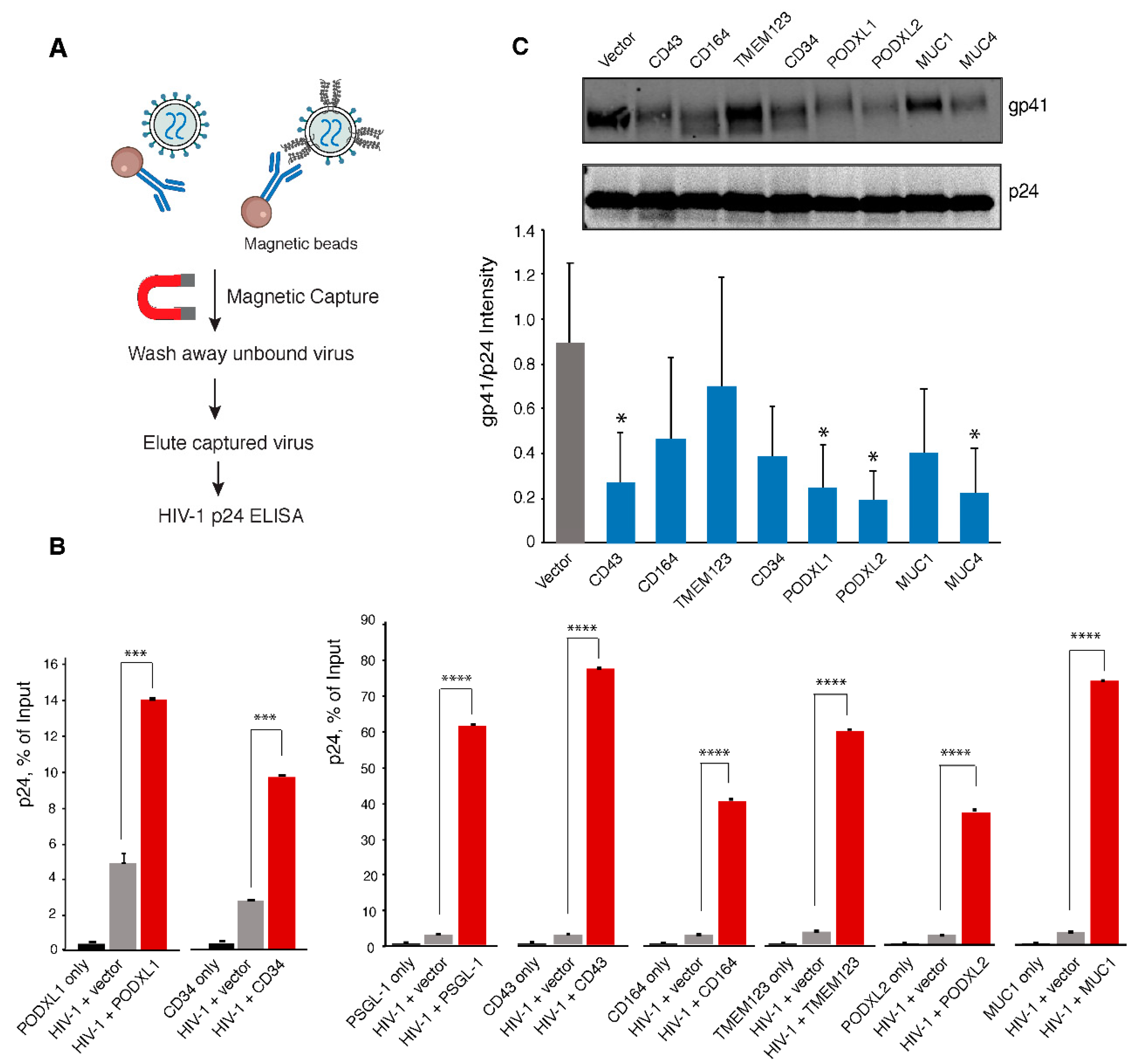
2.6. Viral Attachment Assay
2.7. Western Blots
2.8. p24 ELISA
2.9. Surface Staining
2.10. SARS-CoV-2 S-Antigen ELISA
3. Results
3.1. Inactivation of HIV-1 Virion Infectivity by Mucins and Mucin-Like Proteins
3.2. Effects of SHREK Protein Expression on HIV-1 Virion Release
3.3. Virion Incorporation of SHREK Proteins and Effects on HIV-1 Env Incorporation
3.4. SHREK Proteins Inhibit Virus Particle Attachment to Target Cells
3.5. SHREK Proteins Are Broad-Spectrum Host Antiviral Factors
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hattrup, C.L.; Gendler, S.J. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 2008, 70, 431–457. [Google Scholar] [CrossRef]
- Sako, D.; Chang, X.-J.; Barone, K.M.; Vachino, G.; White, H.M.; Shaw, G.; Veldman, G.M.; Bean, K.M.; Ahern, T.J.; Furie, B.; et al. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell 1993, 75, 1179–1186. [Google Scholar] [CrossRef]
- Goetz, D.J.; Greif, D.M.; Ding, H.; Camphausen, R.T.; Howes, S.; Comess, K.M.; Snapp, K.R.; Kansas, G.S.; Luscinskas, F.W. Isolated P-selectin Glycoprotein Ligand-1 Dynamic Adhesion to P- and E-selectin. J. Cell Biol. 1997, 137, 509–519. [Google Scholar] [CrossRef]
- Guyer, D.A.; Moore, K.L.; Lynam, E.B.; Schammel, C.M.; Rogelj, S.; McEver, R.P.; Sklar, L.A. P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood 1996, 88, 2415–2421. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fu, Y.; Wang, Q.; Li, M.; Zhou, Z.; Dabbagh, D.; Fu, C.; Zhang, H.; Li, S.; Zhang, T.; et al. Proteomic profiling of HIV-1 infection of human CD4+ T cells identifies PSGL-1 as an HIV restriction factor. Nat. Microbiol. 2019, 4, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; He, S.; Waheed, A.A.; Dabbagh, D.; Zhou, Z.; Trinité, B.; Wang, Z.; Yu, J.; Wang, D.; Li, F.; et al. PSGL-1 restricts HIV-1 infectivity by blocking virus particle attachment to target cells. Proc. Natl. Acad. Sci. USA 2020, 117, 9537–9545. [Google Scholar] [CrossRef]
- Murakami, T.; Carmona, N.; Ono, A. Virion-incorporated PSGL-1 and CD43 inhibit both cell-free infection and transinfection of HIV-1 by preventing virus–cell binding. Proc. Natl. Acad. Sci. USA 2020, 117, 8055–8063. [Google Scholar] [CrossRef]
- Li, F.; Erickson, H.P.; James, J.A.; Moore, K.L.; Cummings, R.D.; McEver, R.P. Visualization of P-selectin Glycoprotein Ligand-1 as a Highly Extended Molecule and Mapping of Protein Epitopes for Monoclonal Antibodies. J. Biol. Chem. 1996, 271, 6342–6348. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Moore, K.L.; Cummings, R.D. Leukocyte Trafficking Mediated by Selectin-Carbohydrate Interactions. J. Biol. Chem. 1995, 270, 11025–11028. [Google Scholar] [CrossRef]
- Patel, K.D.; Nollert, M.U.; McEver, R.P. P-selectin must extend a sufficient length from the plasma membrane to mediate rolling of neutrophils. J. Cell Biol. 1995, 131, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wilkins, P.P.; Crawley, S.; Weinstein, J.; Cummings, R.D.; McEver, R.P. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J. Biol. Chem. 1996, 271, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- Pallant, A.; Eskenazi, A.; Mattei, M.G.; Fournier, R.E.; Carlsson, S.R.; Fukuda, M.; Frelinger, J.G. Characterization of cDNAs encoding human leukosialin and localization of the leukosialin gene to chromosome 16. Proc. Natl. Acad. Sci. USA 1989, 86, 1328–1332. [Google Scholar] [CrossRef]
- Umetsu, S.E.; Lee, W.-L.; McIntire, J.J.; Downey, L.; Sanjanwala, B.; Akbari, O.; Berry, G.J.; Nagumo, H.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat. Immunol. 2005, 6, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ablan, S.D.; Miao, C.; Zheng, Y.-M.; Fuller, M.S.; Rennert, P.D.; Maury, W.; Johnson, M.C.; Freed, E.O.; Liu, S.-L. TIM-family proteins inhibit HIV-1 release. Proc. Natl. Acad. Sci. USA 2014, 111, E3699–E3707. [Google Scholar] [CrossRef] [PubMed]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef]
- Nielsen, J.S.; McNagny, K.M. Novel functions of the CD34 family. J. Cell Sci. 2008, 121 Pt 22, 3683–3692. [Google Scholar] [CrossRef]
- Sassetti, C.; Van Zante, A.; Rosen, S.D. Identification of Endoglycan, a Member of the CD34/Podocalyxin Family of Sialomucins. J. Biol. Chem. 2000, 275, 9001–9010. [Google Scholar] [CrossRef] [PubMed]
- Doyonnas, R.; Nielsen, J.S.; Chelliah, S.; Drew, E.; Hara, T.; Miyajima, A.; McNagny, K.M. Podocalyxin is a CD34-related marker of murine hematopoietic stem cells and embryonic erythroid cells. Blood 2005, 105, 4170–4178. [Google Scholar] [CrossRef]
- Watt, S.M.; Chan, J.Y.-H. CD164-A Novel Sialomucin on CD34+Cells. Leuk. Lymphoma 2000, 37, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, C.; Prasad, K.V.S.; Freeman, G.J.; Schlossman, S.F. Molecular cloning of Porimin, a novel cell surface receptor mediating oncotic cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 9778–9783. [Google Scholar] [CrossRef]
- Hetrick, B.; He, S.; Chilin, L.D.; Dabbagh, D.; Alem, F.; Narayanan, A.; Luchini, A.; Li, T.; Liu, X.; Liotta, L.; et al. Development of a novel hybrid alphavirus-SARS-CoV-2 particle for rapid in vitro screening and quantification of neutralization antibodies, antiviral drugs, and viral mutations. bioRxiv 2020. [Google Scholar] [CrossRef]
- Thibault, S.; Tardif, M.R.; Gilbert, C.; Tremblay, M.J. Virus-associated host CD62L increases attachment of human immunodeficiency virus type 1 to endothelial cells and enhances trans infection of CD4+ T lymphocytes. J. Gen. Virol. 2007, 88, 2568–2573. [Google Scholar] [CrossRef]
- Baïsse, B.; Galisson, F.; Giraud, S.; Schapira, M.; Spertini, O. Evolutionary conservation of P-selectin glycoprotein ligand-1 primary structure and function. BMC Evol. Biol. 2007, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Tauxe, C.; Xie, X.; Joffraud, M.; Martinez, M.; Schapira, M.; Spertini, O. P-selectin Glycoprotein Ligand-1 Decameric Repeats Regulate Selectin-dependent Rolling under Flow Conditions. J. Biol. Chem. 2008, 283, 28536–28545. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Beddall, M.H.; Marsh, J.W. Rev-dependent indicator T cell line. Curr. HIV Res. 2007, 5, 394–402. [Google Scholar] [CrossRef]
- Wu, Y.; Beddall, M.H.; Marsh, J.W. Rev-dependent lentiviral expression vector. Retrovirology 2007, 4, 12. [Google Scholar] [CrossRef]
- Barclay, A. Membrane proteins with immunoglobulin-like domains—A master superfamily of interaction molecules. Semin. Immunol. 2003, 15, 215–223. [Google Scholar] [CrossRef]
- Fortin, J.F.; Cantin, R.; Lamontagne, G.; Tremblay, M. Host-derived ICAM-1 glycoproteins incorporated on human immunodeficiency virus type 1 are biologically active and enhance viral infectivity. J. Virol. 1997, 71, 3588–3596. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.F.; Cantin, R.; Tremblay, M.J. T cells expressing activated LFA-1 are more susceptible to infection with human immunodeficiency virus type 1 particles bearing host-encoded ICAM-1. J. Virol. 1998, 72, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Paquette, J.S.; Fortin, J.F.; Blanchard, L.; Tremblay, M.J. Level of ICAM-1 surface expression on virus producer cells influences both the amount of virion-bound host ICAM-1 and human immunodeficiency virus type 1 infectivity. J. Virol. 1998, 72, 9329–9336. [Google Scholar] [CrossRef] [PubMed]
- Tardif, M.R.; Tremblay, M.J. LFA-1 Is a Key Determinant for Preferential Infection of Memory CD4+ T Cells by Human Immunodeficiency Virus Type 1. J. Virol. 2005, 79, 13714–13724. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- McLaren, P.J.; Gawanbacht, A.; Pyndiah, N.; Krapp, C.; Hotter, D.; Kluge, S.F.; Götz, N.; Heilmann, J.; Mack, K.; Sauter, D.; et al. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 2015, 12, 1–15. [Google Scholar] [CrossRef]
- Liu, Y.; Song, Y.; Zhang, S.; Diao, M.; Huang, S.; Li, S.; Tan, X. PSGL-1 inhibits HIV-1 infection by restricting actin dynamics and sequestering HIV envelope proteins. Cell Discov. 2020, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Waheed, A.A.; Hetrick, B.; Dabbagh, D.; Akhrymuk, I.V.; Kehn-Hall, K.; Freed, E.O.; Wu, Y. PSGL-1 Inhibits the Incorporation of SARS-CoV and SARS-CoV-2 Spike Glycoproteins into Pseudovirions and Impairs Pseudovirus Attachment and Infectivity. Viruses 2021, 13, 46. [Google Scholar] [CrossRef]
- Zhou, H.F.; Yan, H.; Cannon, J.L.; Springer, L.E.; Green, J.M.; Pham, C.T. CD43-mediated IFN-gamma production by CD8+ T cells promotes abdominal aortic aneurysm in mice. J. Immunol. 2013, 190, 5078–5085. [Google Scholar] [CrossRef]
- Khunger, A.; Piazza, E.; Warren, S.; Smith, T.H.; Ren, X.; White, A.; Elliott, N.; Cesano, A.; Beechem, J.M.; Kirkwood, J.M.; et al. CTLA-4 blockade and interferon-α induce proinflammatory transcriptional changes in the tumor immune landscape that correlate with pathologic response in melanoma. PLoS ONE 2021, 16, e0245287. [Google Scholar] [CrossRef]
- Rosengren, A.T.; Nyman, T.A.; Syyrakki, S.; Matikainen, S.; Lahesmaa, R. Proteomic and transcriptomic characterization of interferon-?-induced human primary T helper cells. Proteomics 2005, 5, 371–379. [Google Scholar] [CrossRef]
- Reddy, P.K.; Gold, D.V.; Cardillo, T.M.; Goldenberg, D.M.; Li, H.; Burton, J.D. Interferon-gamma upregulates MUC1 expression in haematopoietic and epithelial cancer cell lines, an effect associated with MUC1 mRNA induction. Eur. J. Cancer 2003, 39, 397–404. [Google Scholar] [CrossRef]
- Andrianifahanana, M.; Singh, A.P.; Nemos, C.; Ponnusamy, M.P.; Moniaux, N.; Mehta, P.P.; Varshney, G.C.; Batra, S.K. IFN-gamma-induced expression of MUC4 in pancreatic cancer cells is mediated by STAT-1 upregulation: A novel mechanism for IFN-gamma response. Oncogene 2007, 26, 7251–7261. [Google Scholar] [CrossRef]
- Carter, C.C.; Onafuwa-Nuga, A.; McNamara, L.A.; Riddell, J.; Bixby, D.; Savona, M.R.; Collins, K.L. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 2010, 16, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Habte, H.H.; Kotwal, G.J.; Lotz, Z.E.; Tyler, M.G.; Abrahams, M.; Rodriques, J.; Kahn, D.; Mall, A.S. Antiviral Activity of Purified Human Breast Milk Mucin. Neonatology 2007, 92, 96–104. [Google Scholar] [CrossRef]
- Plante, J.A.; Plante, K.S.; Gralinski, L.E.; Beall, A.; Ferris, M.T.; Bottomly, D.; Green, R.; McWeeney, S.K.; Heise, M.T.; Baric, R.S.; et al. Mucin 4 Protects Female Mice from Coronavirus Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Genet. 2015, 13, 484–496. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabbagh, D.; He, S.; Hetrick, B.; Chilin, L.; Andalibi, A.; Wu, Y. Identification of the SHREK Family of Proteins as Broad-Spectrum Host Antiviral Factors. Viruses 2021, 13, 832. https://doi.org/10.3390/v13050832
Dabbagh D, He S, Hetrick B, Chilin L, Andalibi A, Wu Y. Identification of the SHREK Family of Proteins as Broad-Spectrum Host Antiviral Factors. Viruses. 2021; 13(5):832. https://doi.org/10.3390/v13050832
Chicago/Turabian StyleDabbagh, Deemah, Sijia He, Brian Hetrick, Linda Chilin, Ali Andalibi, and Yuntao Wu. 2021. "Identification of the SHREK Family of Proteins as Broad-Spectrum Host Antiviral Factors" Viruses 13, no. 5: 832. https://doi.org/10.3390/v13050832
APA StyleDabbagh, D., He, S., Hetrick, B., Chilin, L., Andalibi, A., & Wu, Y. (2021). Identification of the SHREK Family of Proteins as Broad-Spectrum Host Antiviral Factors. Viruses, 13(5), 832. https://doi.org/10.3390/v13050832