
High-Throughput Sequencing Indicates Novel Varicosavirus, Emaravirus, and Deltapartitivirus Infections in Vitis coignetiae


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Extraction
2.2. HTS
2.3. Analysis and Comparison of Viral Genome and Encoded Proteins
2.4. RT-PCR
2.5. Inoculation Test
3. Results and Discussion
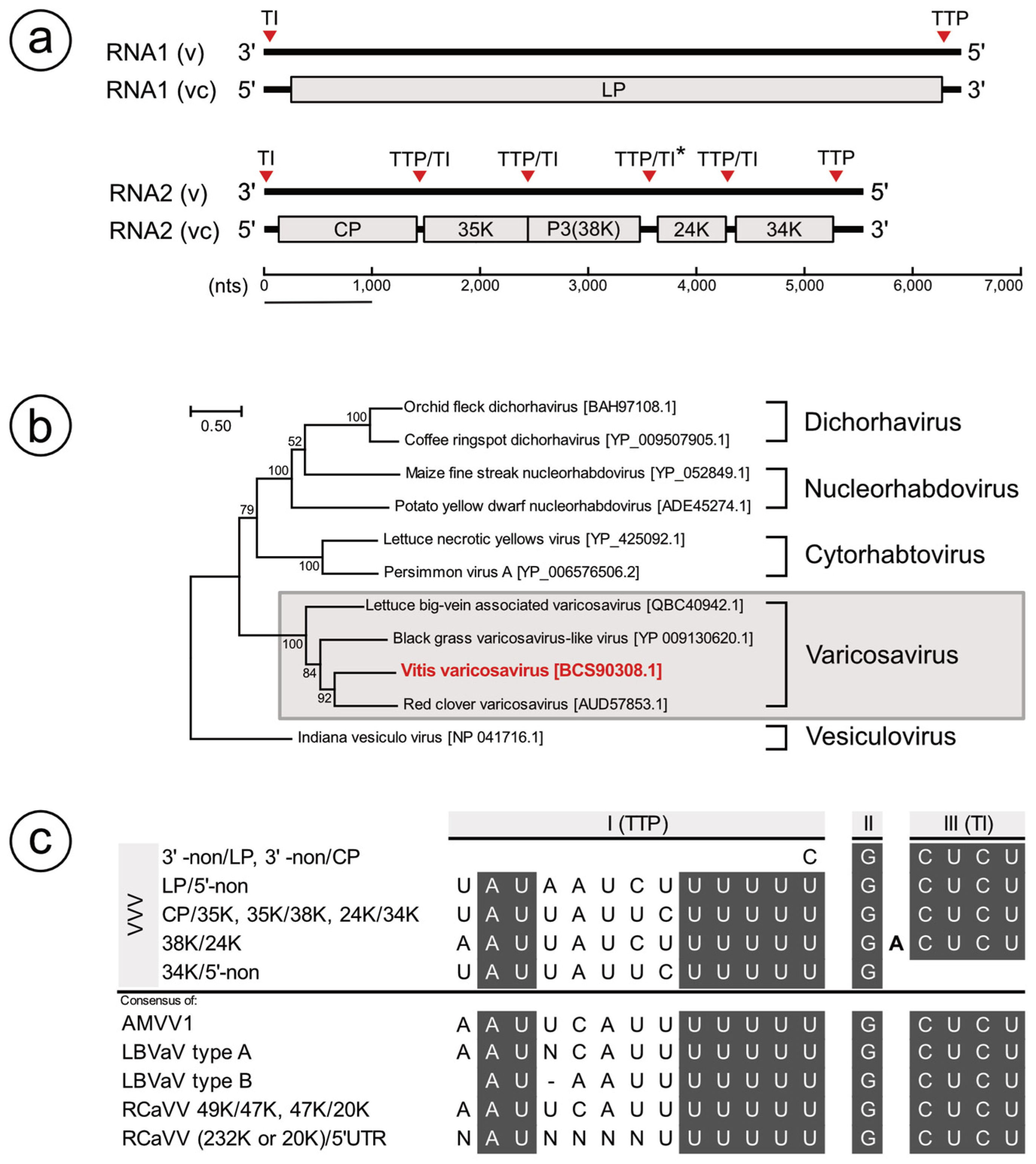
3.1. Identification of a Varicosavirus-Like Virus
3.2. Identification of an Emaravirus-Like Virus
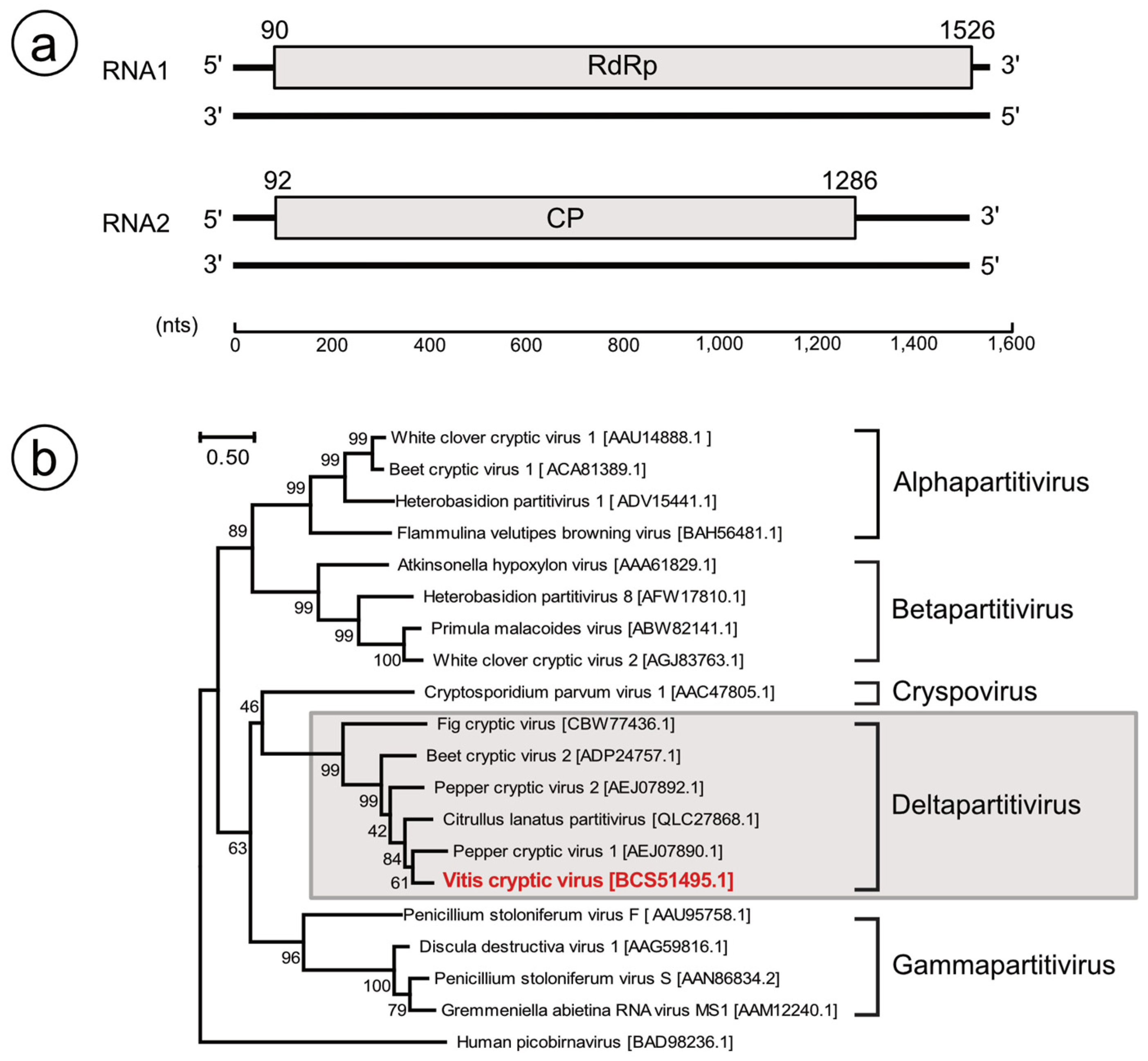
3.3. Identification of Partitivirus-Like Virus
3.4. Mix Infection Status in V. coignetiae
3.5. Inoculation Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.S.; Rumbou, A.; Saldarelli, P.; Škoric, D.; et al. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Martin, R.R.; Constable, F.; Tzanetakis, I.E. Quarantine Regulations and the Impact of Modern Detection Methods. Annu. Rev. Phytopathol. 2016, 54, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Schwaninger, H.R.; Baldo, A.M.; Labate, J.A.; Zhong, G.Y.; Simon, C.J. A phylogenetic analysis of the grape genus (Vitis L.) reveals broad reticulation and concurrent diversification during neogene and quaternary climate change. BMC Evol. Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Umer, M.; Liu, J.; You, H.; Xu, C.; Dong, K.; Luo, N.; Kong, L.; Li, X.; Hong, N.; Wang, G.; et al. Genomic, morphological and biological traits of the viruses infecting major fruit trees. Viruses 2019, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, G.; Ueki, K.; Ichi, T.; Aoki, H.; Fujiwara, M.; Hirano, K. Juice constituents and skin pigments in Vitis coignetiae Pulliat grapevines. Vitis 2002, 41, 161–162. [Google Scholar]
- Yun, J.W.; Lee, W.S.; Kim, M.J.; Lu, J.N.; Kang, M.H.; Kim, H.G.; Kim, D.C.; Choi, E.J.; Choi, J.Y.; Kim, H.G.; et al. Characterization of a profile of the anthocyanins isolated from Vitis coignetiae Pulliat and their anti-invasive activity on HT-29 human colon cancer cells. Food Chem. Toxicol. 2010, 48, 903–909. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef]
- Verbeek, M.; Dullemans, A.M.; van Bekkum, P.J.; van der Vlugt, R.A.A. Evidence for Lettuce big-vein associated virus as the causal agent of a syndrome of necrotic rings and spots in lettuce. Plant Pathol. 2013, 62, 444–451. [Google Scholar] [CrossRef]
- Sabbadin, F.; Glover, R.; Stafford, R.; Rozado-Aguirre, Z.; Boonham, N.; Adams, I.; Mumford, R.; Edwards, R. Transcriptome sequencing identifies novel persistent viruses in herbicide resistant wild-grasses. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Koloniuk, I.; Fránová, J.; Sarkisova, T.; Přibylová, J.; Lenz, O.; Petrzik, K.; Špak, J. Identification and molecular characterization of a novel varicosa-like virus from red clover. Arch. Virol. 2018, 163, 2213–2218. [Google Scholar] [CrossRef]
- Sasaya, T.; Kusaba, S.; Ishikawa, K.; Koganezawa, H. Nucleotide sequence of RNA2 of Lettuce big-vein virus and evidence for a possible transcription termination/initiation strategy similar to that of rhabdoviruses. J. Gen. Virol. 2004, 85, 2709–2717. [Google Scholar] [CrossRef]
- Rose, J.K. Complete intergenic and flanking gene sequences from the genome of vesicular stomatitis virus. Cell 1980, 19, 415–421. [Google Scholar] [CrossRef]
- Elbeaino, T.; Digiaro, M.; Mielke-Ehret, N.; Muehlbach, H.P.; Martelli, G.P. ICTV virus taxonomy profile: Fimoviridae. J. Gen. Virol. 2018, 99, 1478–1479. [Google Scholar] [CrossRef]
- Mielke, N.; Muehlbach, H.P. A novel, multipartite, negative-strand RNA virus is associated with the ringspot disease of European mountain ash (Sorbus aucuparia L.). J. Gen. Virol. 2007, 88, 1337–1346. [Google Scholar] [CrossRef]
- Mielke-Ehret, N.; Mühlbach, H.P. Emaravirus: Anovel genus of multipartite, negative strand RNA plant viruses. Viruses 2012, 4, 1515–1536. [Google Scholar] [CrossRef] [PubMed]
- von Bargen, S.; Al Kubrusli, R.; Gaskin, T.; Fürl, S.; Hüttner, F.; Blystad, D.R.; Karlin, D.G.; Jalkanen, R.; Büttner, C. Characterisation of a novel Emaravirus identified in mosaic-diseased Eurasian aspen (Populus tremula). Ann. Appl. Biol. 2020, 176, 210–222. [Google Scholar] [CrossRef]
- Kumar, S.; Subbarao, B.; Hallan, V. Molecular characterization of emaraviruses associated with Pigeonpea sterility mosaic disease. Sci. Rep. 2017, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Elbeaino, T.; Digiaro, M.; Alabdullah, A.; De Stradis, A.; Minafra, A.; Mielke, N.; Castellano, M.A.; Martelli, G.P. A multipartite single-stranded negative-sense RNA virus is the putative agent of fig mosaic disease. J. Gen. Virol. 2009, 90, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Di Bello, P.L.; Keller, K.E.; Martin, R.R.; Sabanadzovic, S.; Tzanetakis, I.E. A new, widespread emaravirus discovered in blackberry. Virus Res. 2017, 235, 1–5. [Google Scholar] [CrossRef]
- Walia, J.J.; Salem, N.M.; Falk, B.W. Partial sequence and survey analysis identify a multipartite, negative-sense RNA virus associated with fig mosaic. Plant Dis. 2009, 93, 4–10. [Google Scholar] [CrossRef]
- Zheng, Y.; Navarro, B.; Wang, G.; Wang, Y.; Yang, Z.; Xu, W.; Zhu, C.; Wang, L.; Di Serio, F.; Hong, N. Actinidia chlorotic ringspot-associated virus: A novel emaravirus infecting kiwifruit plants. Mol. Plant Pathol. 2017, 18, 569–581. [Google Scholar] [CrossRef]
- Buzkan, N.; Chiumenti, M.; Massart, S.; Sarpkaya, K.; Karadağ, S.; Minafra, A. A new emaravirus discovered in Pistacia from Turkey. Virus Res. 2019, 263, 159–163. [Google Scholar] [CrossRef]
- Tatineni, S.; McMechan, A.J.; Wosula, E.N.; Wegulo, S.N.; Graybosch, R.A.; French, R.; Hein, G.L. An Eriophyid Mite-Transmitted Plant Virus Contains Eight Genomic RNA Segments with Unusual Heterogeneity in the Nucleocapsid Protein. J. Virol. 2014, 88, 11834–11845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, Y.; Cao, M.; Cheng, Q.; Wu, J.; Hu, T. Identification of a novel emaravirus infecting lilac through next-generation sequencing. J. Integr. Agric. 2020, 19, 2064–2071. [Google Scholar] [CrossRef]
- Buck, K.W.; Kempson-Jones, F. Biophysical Properties of Penicillium stoloniferum Virus S. J. Virol. 1973, 18, 223–235. [Google Scholar] [CrossRef]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M.; Simmonds, P.; et al. ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Nibert, M.L.; Ghabrial, S.A.; Maiss, E.; Lesker, T.; Vainio, E.J.; Jiang, D.; Suzuki, N. Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res. 2014, 188, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Woods, K.M.; Upton, S.J.; Ghabrial, S.A. Cryspovirus: A new genus of protozoan viruses in the family Partitiviridae. Arch. Virol. 2009, 154, 1959–1965. [Google Scholar] [CrossRef]
- Xin, M.; Cao, M.; Liu, W.; Ren, Y.; Lu, C.; Wang, X. The genomic and biological characterization of Citrullus lanatus cryptic virus infecting watermelon in China. Virus Res. 2017, 232, 106–112. [Google Scholar] [CrossRef]
- Sela, N.; Lachman, O.; Reingold, V.; Dombrovsky, A. A new cryptic virus belonging to the family Partitiviridae was found in watermelon co-infected with Melon necrotic spot virus. Virus Genes 2013, 47, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.J.; Osman, T.A.M.; Booy, F.P.; Coutts, R.H.A.; Brasier, C.M.; Buck, K.W. Molecular characterization of a partitivirus from Ophiostoma himal-ulmi. Virus Genes 2006, 33, 33–39. [Google Scholar] [CrossRef]
- Kim, H.; Park, D.; Hahn, Y. Identification of novel RNA viruses in alfalfa (Medicago sativa): An Alphapartitivirus, a Deltapartitivirus, and a Marafivirus. Gene 2018, 638, 7–12. [Google Scholar] [CrossRef]
- Abe, J.; Nabeshima, T. First report of grapevine Pinot gris virus in wild grapevines (Vitis coignetiae) in Japan. J. Plant Pathol. 2021, in press. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Roumi, V.; Roberto, R.; Malossini, U.; Yoshikawa, N.; La Notte, P.; Terlizzi, F.; Credi, R.; Saldarelli, P. A new grapevine virus discovered by deep sequencing of virus- and viroid-derived small RNAs in Cv Pinot gris. Virus Res. 2012, 163, 262–268. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Iida, H.; Goto, S.; Magome, H.; Takahashi, T.; Terai, Y. Grapevine berry inner necrosis, a new trichovirus: Comparative studies with several known trichoviruses. Arch. Virol. 1997, 142, 1351–1363. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Bar-Joseph, M.; Brunt, A.A.; Candresse, T.; Foster, G.D.; Martelli, G.P.; Milne, R.G.; Fauquet, C.M. The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch. Virol. 2004, 149, 1045–1060. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pool | Sample No. | Location (Prefecture) | Symptom | Viral Infection a | ||||
|---|---|---|---|---|---|---|---|---|
| VVV | VEV | VCV | GPGV | GINV | ||||
| A | 1 | 45°07′46.59″ N, 142°12′06.04″ E (Hokkaido) | Mosaic | + | − | − | + | + |
| 2 | 45°07′44.85″ N, 142°12′05.18″ E (Hokkaido) | None | − | − | + | + | + | |
| 4 | 45°07′42.63″ N, 142°12′02.99″ E (Hokkaido) | None | − | + | + | − | − | |
| 5 | 45°07′41.91″ N, 142°12′02.86″ E (Hokkaido) | None | − | + | + | − | − | |
| 6 | 45°07′40.39″ N, 142°12′02.21″ E (Hokkaido) | None | − | + | + | − | − | |
| 7 | 45°07′43.23″ N, 142°12′03.69″ E (Hokkaido) | Mosaic | − | − | − | + | + | |
| 8 | 45°07′46.74″ N, 142°12′03.09″ E (Hokkaido) | Mosaic | + | − | − | + | + | |
| 9 | 45°07′49.68″ N, 142°11′24.80″ E (Hokkaido) | None | − | − | + | + | + | |
| 11 | 44°57′27.96″ N, 142°13′19.85″ E (Hokkaido) | None | − | − | + | − | − | |
| 12 | 44°52′55.93″ N, 142°12′43.34″ E (Hokkaido) | None | − | − | − | − | − | |
| 13 | 44°56′25.57″ N, 142°13′28.72″ E (Hokkaido) | None | − | − | − | − | − | |
| 14 | 44°56′15.11″ N, 142°13′18.44″ E (Hokkaido) | None | − | − | − | − | − | |
| 15 | 44°53′35.22″ N, 142°12′10.86″ E (Hokkaido) | None | − | − | − | − | − | |
| 16 | 44°53′33.27″ N, 142°12′06.99″ E (Hokkaido) | None | − | − | − | − | − | |
| 17 | 44°53′30.92″ N, 142°12′07.58″ E (Hokkaido) | None | − | + | + | − | − | |
| B | 18 | 43°08′03.04″ N, 140°49′27.85″ E (Hokkaido) | None | − | − | + | − | − |
| 19 | 43°08′00.95″ N, 140°49′30.55″ E (Hokkaido) | None | − | + | + | − | − | |
| 20 | 43°00′28.97″ N, 140°52′34.70″ E (Hokkaido) | None | − | − | − | − | − | |
| 21 | 43°00′23.97″ N, 140°52′41.01″ E (Hokkaido) | None | − | − | − | − | − | |
| 22 | 43°21′23.14″ N, 142°14′37.98″ E (Hokkaido) | None | − | − | − | − | − | |
| 23 | 43°21′18.17″ N, 142°14′35.64″ E (Hokkaido) | None | − | − | + | − | − | |
| 24 | 43°27′19.82″ N, 142°30′06.96″ E (Hokkaido) | None | − | − | + | − | − | |
| 25 | 43°27′19.88″ N, 142°30′14.23″ E (Hokkaido) | None | − | − | − | − | − | |
| 26 | 43°26′21.10″ N, 142°35′43.27″ E (Hokkaido) | None | − | + | + | − | − | |
| 30 | 40°11′22.55″ N, 141°29′15.77″ E (Iwate) | Leaf roll | − | − | + | − | + | |
| 31 | 40°07′45.35″ N, 141°31′08.13″ E (Iwate) | Leaf roll | − | − | + | − | + | |
| 32 | 40°07′44.72″ N, 141°31′04.26″ E (Iwate) | Leaf roll | − | − | + | − | − | |
| 33 | 40°07′45.17″ N, 141°30′59.01″ E (Iwate) | Mosaic | − | − | + | − | + | |
| 34 | 39°00′18.48″ N, 140°51′42.33″ E (Iwate) | None | − | − | + | − | − | |
| 35 | 39°00′15.42″ N, 140°51′35.98″ E (Iwate) | None | − | − | + | − | − | |
| 36 | 39°59′03.19″ N, 140°43′08.54″ E (Akita) | None | − | − | − | − | − | |
| 37 | 38°58′51.20″ N, 140°43′04.73″ E (Akita) | None | − | − | + | − | − | |
| 38 | 39°00′55.83″ N, 140°38′36.00″ E (Akita) | None | − | − | + | − | − | |
| Virus a | Genomic Segments | Contig No. b | Length (nt) | No. of Reads | Best Much in BlastX | Query Coverage (%) | Sequence Identity (%) | Accession Numbers (NCBI, GenBank) |
|---|---|---|---|---|---|---|---|---|
| VVV | RNA1 | 38A | 6450 | 19,629 | Lettuce big-vein associated varicosavirus [QBC40942.1] | 90 | 39.6 | LC604719 |
| RNA2 | 85A | 5535 | 20,846 | Black grass varicosavirus-like virus [YP009130617.1] | 16 | 27.0 | LC604720 | |
| VEV | RNA1a | 201A | 7083 | 11,122 | Aspen mosaic-associated virus [CAA0079389.1] | 96 | 49.3 | LC604721 |
| RNA2a | 69A | 2099 | 20,739 | Pigeonpea sterility mosaic emaravirus 2 [QBA83608.1] | 91 | 43.1 | LC604722 | |
| RNA3a | 19A | 1209 | 7735 | Fig mosaic emaravirus [AWS21340.1] | 74 | 44.1 | LC604723 | |
| RNA3b | 574A | 881 | 308 | Fig mosaic emaravirus [AWS21340.1] | 91 | 44.4 | LC604732 | |
| RNA4a | 184A | 1270 | 574 | Pigeonpea sterility mosaic emaravirus 2 [ANQ90780.1] | 84 | 39.8 | LC604724 | |
| RNA4b | 185A | 1596 | 8948 | Pigeonpea sterility mosaic emaravirus 2 [ANQ90780.1] | 67 | 39.8 | LC604725 | |
| RNA6a | 70A | 1335 | 13,473 | Pistacia emaravirus [QAR18008.1] | 42 | 25.5 | LC604726 | |
| RNA1b | 311B | 7053 | 1149 | Aspen mosaic-associated virus [CAA0079389.1] | 97 | 48.9 | LC604727 | |
| RNA2b | 179B | 2091 | 1249 | Pigeonpea sterility mosaic emaravirus 2 [QBA83608.1] | 91 | 43.0 | LC604728 | |
| RNA3c | 58B | 1211 | 1149 | Fig mosaic emaravirus [AWS21340.1] | 74 | 44.1 | LC604729 | |
| RNA4c | 76B | 1628 | 2137 | Pigeonpea sterility mosaic emaravirus 2 [ANQ90780.1] | 65 | 39.5 | LC604730 | |
| RNA6b | 123B | 1324 | 3090 | Pistacia emaravirus [QAR18008.1] | 38 | 25.3 | LC604731 | |
| VCV | RNA1a | 369A | 1563 | 1843 | Citrullus lanatus cryptic virus [APT68925.1] | 90 | 66.7 | LC602838 |
| RNA2a | 784A | 1521 | 937 | Citrullus lanatus partitivirus [QLC27869.1] | 77 | 47.0 | LC602839 | |
| RNA1b | 567B | 1563 | 1349 | Citrullus lanatus cryptic virus [APT68925.1] | 90 | 65.6 | LC602840 | |
| RNA2b | 146B | 1521 | 746 | Citrullus lanatus partitivirus [QLC27869.1] | 77 | 47.3 | LC602841 |
| Vitis Varicosavirus (VVV) | |||
|---|---|---|---|
| LP a | CP a | P3 a | |
| Red clover varicosavirus (RCaVV) | 40.9 | 30.1 | - |
| Lettuce big-vein-associated varicosavirus (LBVaV) | 39.6 | 27.3 | 29.0 |
| Alopecurus myosuroides varicosavirus 1 (AMVV1) | 38.6 | 27.0 | - |
| Virus | Vitis Emaravirus (VEV) | ||||
|---|---|---|---|---|---|
| RdRp [GenBank] | GP [GenBank] | CP [GenBank] | MP [GenBank] | P6 [GenBank] | |
| Pistacia emaravirus | 48.9 [QAR18002.1] | 39.3 [QAR18003.1] | 38.6 [QAR18004.1] | 35.0 [QAR18005.1] | 22.6 [QAR18008.1] |
| Pigeonpea sterility mosaic emaravirus 2 | 48.6 [CCV01186.1] | 42.1 [YP_009268865.1] | 40.3 [YP_009268864.1] | 37.4 [YP_009268866.1] | 21.0 [YP_009268862.1] |
| Pigeonpea sterility mosaic emaravirus 1 | 48.3 [CCP46989.1] | 39.7 [YP_009237263.1] | 38.0 [YP_009237281.1] | - a | - a |
| Blackberry leaf mottle-associated virus | 48.2 [AQX45473.1] | 40.1 [AQX45474.1] | 38.3 [AQX45475.1] | 37.4 [AQX45476.1] | - a |
| Redbud yellow ringspot-associated emaravirus | 48.0 [AEO95760.1] | 39.2 [YP_009508087.1] | 32.7 [YP_009508085.1] | 23.4 [YP_009508084.1] | - a |
| Fig mosaic emaravirus | 48.0 [CAQ03479.7] | 40.8 [YP_009237272.1] | 40.5 [YP_009237270.1] | 36.3 [YP_009237271.1] | 14.3 [YP_009237275.1] |
| European mountain ash ringspot-associated emaravirus | 47.9 [AAS73287.2] | 36.4 [YP_003104765.1] | 36.6 [YP_003104767.1] | 7.7 [YP_003104766.1] | - a |
| Rose rosette emaravirus | 47.9 [ADZ54688.1] | 41.2 [YP_004327590.1] | 35.8 [YP_004327591.1] | 36.6 [YP_004327592.1] | 20.8 [YP_009380548.1] |
| Actinidia chlorotic ringspot-associated virus | 47.8 [ALX00127.1] | 37.8 [YP_009507926.1] | 34.6 [YP_009507928.1] | 25.1 [YP_009507927.1] | - a |
| Raspberry leaf blotch emaravirus | 32.4 [CBZ42024.1] | 21.0 [YP_009237265.1] | 18.7 [YP_009237266.1] | 17.6 [YP_009237267.1] | 11.6 [YP_009237278.1] |
| High Plains wheat mosaic emaravirus | 30.2 [AIK23031.1] | 21.0 [YP_009237256.1] | 18.3 [YP_009237257.1] | 18.6 [YP_009237258.1] | 11.4 [YP_009237260.1] |
| Genus | Virus | Vitis Cryptic Virus (Pool A;BCS51495.1/BCS51469.1) | |
|---|---|---|---|
| RdRp [GenBank] | CP [GenBank] | ||
| Deltapartitivirus | Vitis cryptic virus (Pool B) | 95.8 [BCS51497.1] | 98.2 [BCS51498.1] |
| Citrulluslanatus partitivirus | 66.7 [APT68925.1] | 47.5 [QLC27869.1] | |
| Pepper cryptic virus 1 | 65.7 [QEO60284.1] | 39.7 [AVV48359.1] | |
| Pepper cryptic virus 2 | 59.9 [AVL84364.1] | 37.9 [ALR34989.1] | |
| Beet cryptic virus 2 | 58.6 [QCF59322.1] | 32.6 [QCF59321.1] | |
| Fig cryptic virus | 37.7 [CBZ05548.1] | 18.7 [YP_004429259.1] | |
| Alphapartitivirus | White clover cryptic virus 1 | 19.6 [YP_086754.1] | 4.5 [YP_086755.1] |
| Betapartitivirus | Atkinsonella hypoxylon virus | 18.1 [NP_604475.1] | 14.0 [NP_604476.1] |
| Crypsovirus | Cryptosporidium parvum virus 1 | 21.3 [YP_009508065.1] | 16.8 [YP_009508066.1] |
| Gammapartitivirus | Penicillium stoloniferum virus S | 19.7 [YP_052856.2] | 1.8 [YP_052857.1] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nabeshima, T.; Abe, J. High-Throughput Sequencing Indicates Novel Varicosavirus, Emaravirus, and Deltapartitivirus Infections in Vitis coignetiae. Viruses 2021, 13, 827. https://doi.org/10.3390/v13050827
Nabeshima T, Abe J. High-Throughput Sequencing Indicates Novel Varicosavirus, Emaravirus, and Deltapartitivirus Infections in Vitis coignetiae. Viruses. 2021; 13(5):827. https://doi.org/10.3390/v13050827
Chicago/Turabian StyleNabeshima, Tomoyuki, and Junya Abe. 2021. "High-Throughput Sequencing Indicates Novel Varicosavirus, Emaravirus, and Deltapartitivirus Infections in Vitis coignetiae" Viruses 13, no. 5: 827. https://doi.org/10.3390/v13050827
APA StyleNabeshima, T., & Abe, J. (2021). High-Throughput Sequencing Indicates Novel Varicosavirus, Emaravirus, and Deltapartitivirus Infections in Vitis coignetiae. Viruses, 13(5), 827. https://doi.org/10.3390/v13050827