
The Development of Bacteriophage Resistance in Vibrio alginolyticus Depends on a Complex Metabolic Adaptation Strategy

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Vibrio alginolyticus Strain V1
2.2. Bacteriophages Aphrodite1, phiSt2, and Ares1
2.3. Vibrio alginolyticus Phage-Resistant Strains
2.4. Growth Kinetics, Phage Biological Characteristics, and Phage Adsorption Dynamics
2.5. Phage DNA Extraction and Sequencing
2.6. Bacterial DNA Extraction and Sequencing
2.7. Bacteria and Bacteriophage Genome Analyses
2.8. Gene Expression Analysis
2.9. Metabolite Extraction and Intracellular Metabolite Analysis
2.10. Statistical Analysis
2.11. Accession Numbers
3. Results
3.1. Description of Novel Bacteriophage Isolates
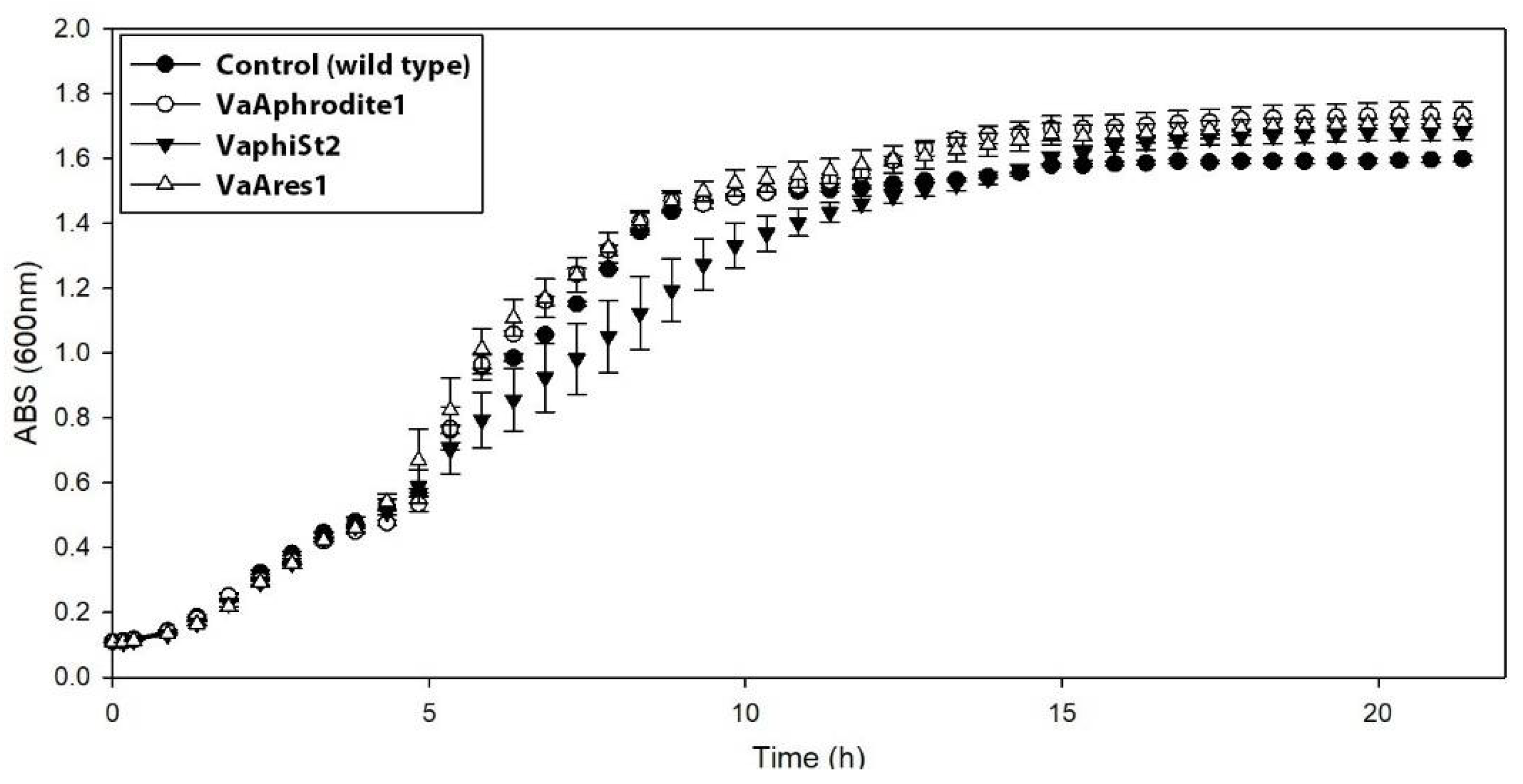
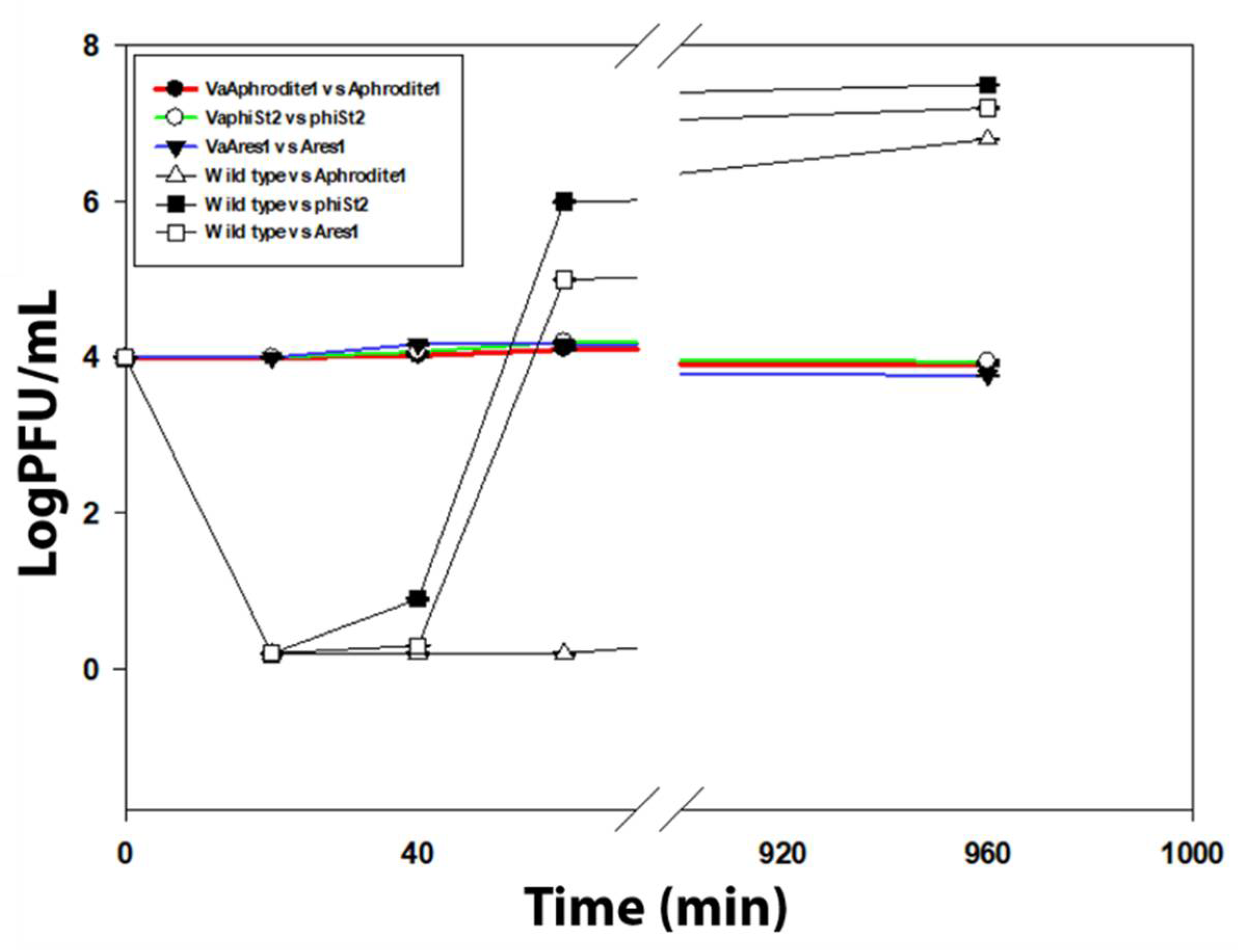
3.2. Cross Infection and Growth Curve of Phage-Resistant Strains
3.3. Study of Anti-Phage Defense Systems of Vibrio alginolyticius
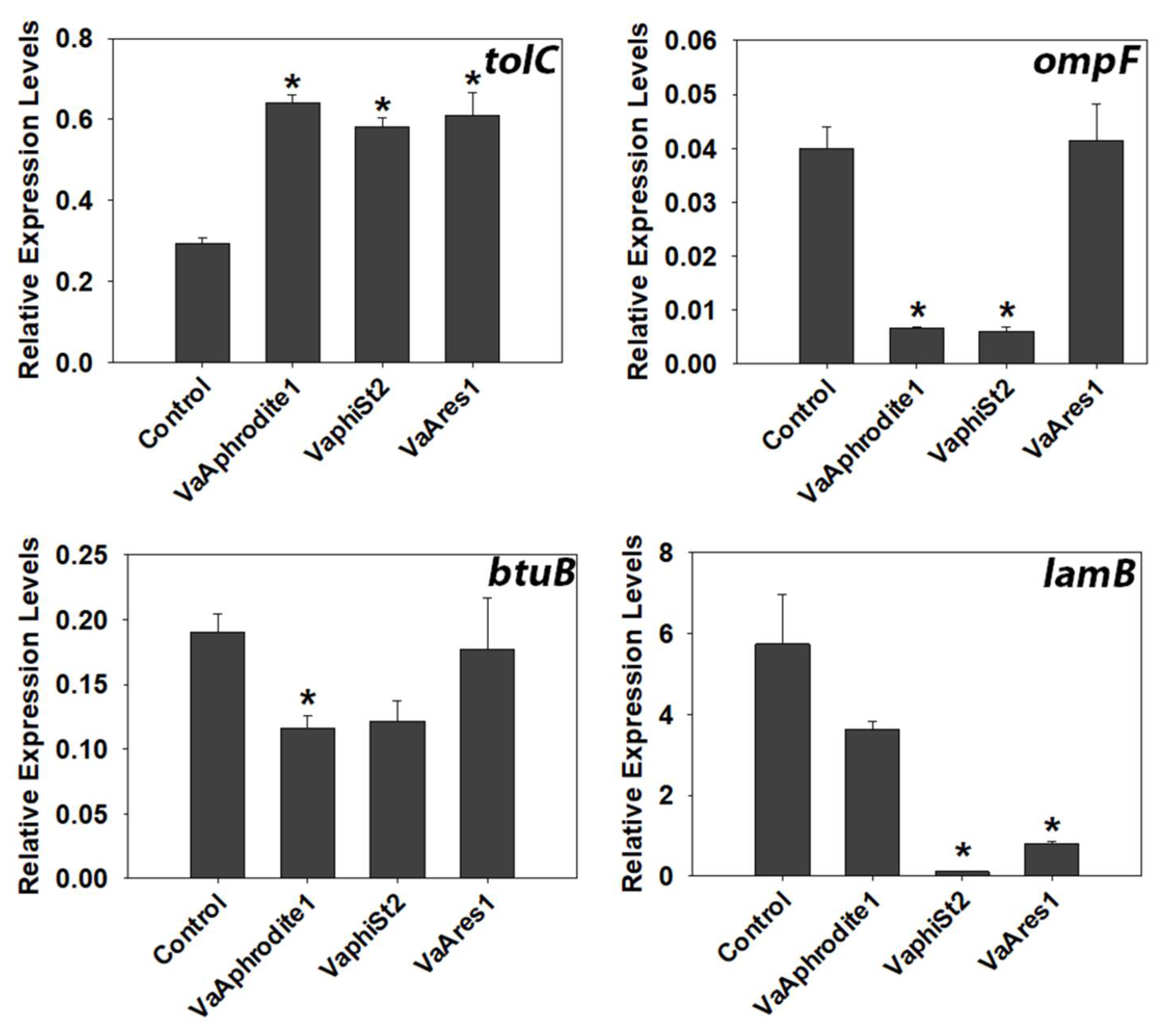
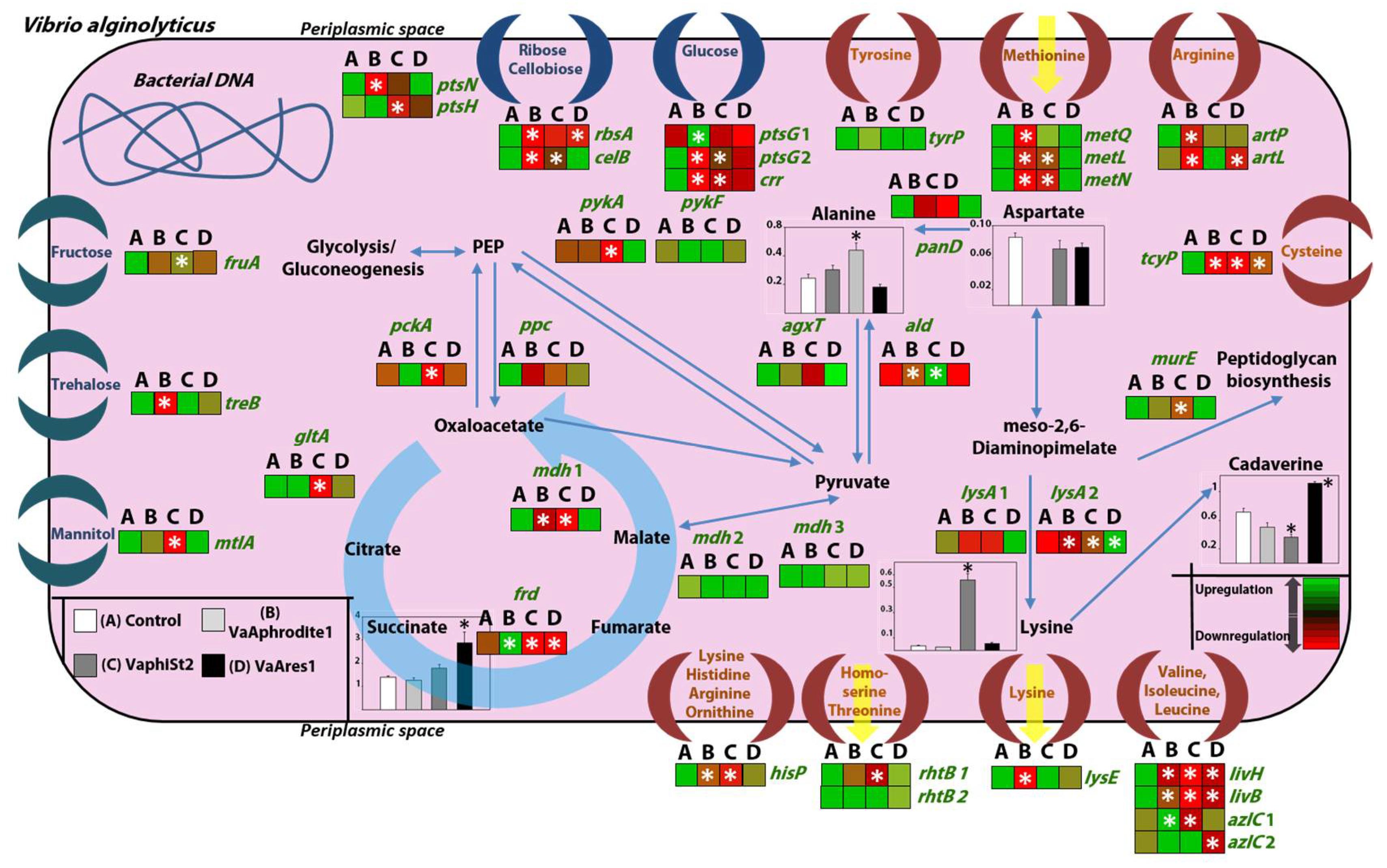
3.4. Transcript Profiling of Bacterial Genes Involved in the Phage Infection Process
3.5. Transcript Profiling of Genes Coding for Key Metabolic Enzymes
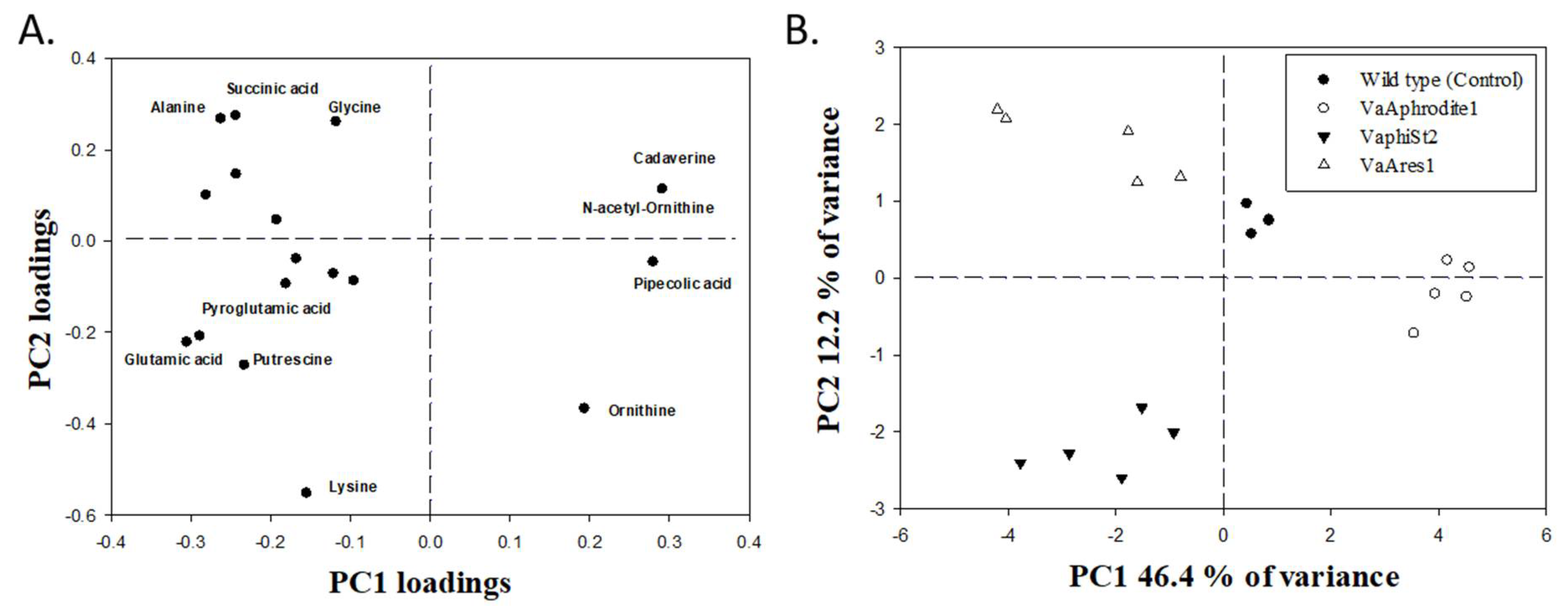
3.6. Metabolomic Profiling of the Resistant Strains
3.7. Comparative Genomic Analysis of Resistant Vibrio Strains
4. Discussion
4.1. Major Transcriptional Reprogramming Could Confer Phage Tolerance
4.2. Resistance to Lytic Bacteriophages Includes Metabolic Adaptation Mechanisms
4.3. Transcription Reprogramming in Phage-Resistant Becterial Strains Could Be Triggered by Genomic Lesions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lennon, J.T.; Khatana, S.A.M.; Marston, M.F.; Martiny, J.B.H. Is there a cost of virus resistance in marine cyanobacteria? ISME J. 2007, 1, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Buckling, A.; Rainey, P.B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. R. Soc. B Biol. Sci. 2002, 269, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Baptista, C.; Santos, M.A.; São-José, C. Phage SPP1 Reversible Adsorption to Bacillus subtilis Cell Wall Teichoic Acids Accelerates Virus Recognition of Membrane Receptor YueB. J. Bacteriol. 2008, 190, 4989–4996. [Google Scholar] [CrossRef]
- São-José, C.; De Frutos, M.; Raspaud, E.; Santos, M.A.; Tavares, P. Pressure built by DNA packing inside virions: Enough to drive DNA ejection in vitro, largely insufficient for delivery into the bacterial cytoplasm. J. Mol. Biol. 2007, 374, 346–355. [Google Scholar] [CrossRef]
- Jakutytė, L.; Baptista, C.; São-José, C.; Daugelavičius, R.; Lopez, R.C.; Tavares, P. Bacteriophage Infection in Rod-Shaped Gram-Positive Bacteria: Evidence for a Preferential Polar Route for Phage SPP1 Entry in Bacillus subtilis. J. Bacteriol. 2011, 193, 4893–4903. [Google Scholar] [CrossRef]
- Rakhuba, D.V.; Kolomiets, E.I.; Dey, E.S.; Novik, G.I. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol. J. Microbiol. 2010, 59, 145–155. [Google Scholar] [CrossRef]
- Oliveira, H.; Thiagarajan, V.; Walmagh, M.; Sillankorva, S.; Lavigne, R.; Neves-Petersen, M.T.; Kluskens, L.D.; Azeredo, J. A Thermostable Salmonella Phage Endolysin, Lys68, with Broad Bactericidal Properties against Gram-Negative Pathogens in Presence of Weak Acids. PLoS ONE 2014, 9, e108376. [Google Scholar] [CrossRef]
- Grayson, P.; Molineux, I.J. Is phage DNA “injected” into cells—Biologists and physicists can agree. Curr. Opin. Microbiol. 2007, 10, 401–409. [Google Scholar] [CrossRef][Green Version]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef]
- Tarahovsky, Y.S.; Khusainov, A.A.; Deev, A.A.; Kim, Y.V. Membrane fusion during infection of Escherichia coli cells by phage T4. FEBS Lett. 1991, 289, 8–22. [Google Scholar] [CrossRef]
- Tarahovsky, Y.; Khusainov, A.; Daugelavichus, R.; Bakene, E. Structural changes in Escherichia coli membranes induced by bacteriophage T4 at different temperatures. Biophys. J. 1995, 68, 157–163. [Google Scholar] [CrossRef][Green Version]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928. [Google Scholar] [CrossRef]
- Bonhivers, M.; Ghazi, A.; Boulanger, P.; Letellier, L. FhuA, a transporter of the Escherichia coli outer membrane, is converted into a channel upon binding of bacteriophage T5. EMBO J. 1996, 15, 1850–1856. [Google Scholar] [CrossRef]
- Chatterjee, S.; Rothenberg, E. Interaction of Bacteriophage l with Its E. coli Receptor, LamB. Viruses 2012, 4, 3162–3178. [Google Scholar] [CrossRef]
- Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef]
- Barabote, R.D.; Saier, M.H. Comparative Genomic Analyses of the Bacterial Phosphotransferase System. Microbiol. Mol. Biol. Rev. 2005, 69, 608–634. [Google Scholar] [CrossRef]
- Siebold, C.; Flükiger, K.; Beutler, R.; Erni, B. Carbohydrate transporters of the bacterial phosphoenolpyruvate: Sugar phosphotransferase system (PTS). FEBS Lett. 2001, 504, 104–111. [Google Scholar] [CrossRef]
- Davidson, A.L.; Maloney, P.C. ABC transporters: How small machines do a big job. Trends Microbiol. 2007, 15, 448–455. [Google Scholar] [CrossRef]
- Holmfeldt, K.; Middelboe, M.; Nybroe, O.; Riemann, L. Large Variabilities in Host Strain Susceptibility and Phage Host Range Govern Interactions between Lytic Marine Phages and Their Flavobacterium Hosts. Appl. Environ. Microbiol. 2007, 73, 6730–6739. [Google Scholar] [CrossRef]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nat. Cell Biol. 2020, 577, 327–336. [Google Scholar] [CrossRef]
- Bernheim, A.; Sorek, R. The pan-immune system of bacteria: Antiviral defence as a community resource. Nat. Rev. Genet. 2020, 18, 113–119. [Google Scholar] [CrossRef]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR—A widespread system that pro-vides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2014, 34, 169–183. [Google Scholar] [CrossRef]
- Swarts, D.C.; Jore, M.M.; Westra, E.R.; Zhu, Y.; Janssen, J.H.; Snijders, A.P.; Wang, Y.; Patel, D.J.; Berenguer, J.; Brouns, S.J.J.; et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nat. Cell Biol. 2014, 507, 258–261. [Google Scholar] [CrossRef]
- Ofir, G.; Melamed, S.; Sberro, H.; Mukamel, Z.; Silverman, S.; Yaakov, G.; Doron, S.; Sorek, R. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat. Microbiol. 2018, 3, 90–98. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.G.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 1–13. [Google Scholar] [CrossRef]
- Mustapha, S.; Mustapha, E.M.; Nozha, C. Vibrio alginolyticus: An emerging pathogen of food borne diseases. Int. J. Sci. Technol. 2013, 2, 302–309. [Google Scholar]
- Castillo, D.; D’Alvise, P.; Kalatzis, P.G.; Kokkari, C.; Middelboe, M.; Gram, L.; Liu, S.; Katharios, P. Draft Genome Sequences ofVibrio alginolyticusStrains V1 and V2, Opportunistic Marine Pathogens. Genome Announc. 2015, 3, e00729-15. [Google Scholar] [CrossRef]
- Kalatzis, P.G.; Bastías, R.; Kokkari, C.; Katharios, P. Isolation and Characterization of Two Lytic Bacteriophages, phiSt2 and phiGrn1; Phage Therapy Application for Biological Control of Vibrio alginolyticus in Aquaculture Live Feeds. PLoS ONE 2016, 11, e0151101. [Google Scholar] [CrossRef]
- Skliros, D.; Kalatzis, P.G.; Katharios, P.; Flemetakis, E. Comparative Functional Genomic Analysis of Two Vibrio Phages Reveals Complex Metabolic Interactions with the Host Cell. Front. Microbiol. 2016, 7, 1807. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E. Phage host range and efficiency of plating. Methods Mol. Biol. 2009, 501, 141–149. [Google Scholar] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Chai, G.; Yu, M.; Jiang, L.; Duan, Y.; Huang, J. HMMCAS: A Web Tool for the Identification and Domain Annotations of CAS Proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1313–1315. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Lekanne Deprez, R.H.; Moorman, A.F.M. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Maharjan, R.P.; Ferenci, T. Global metabolite analysis: The influence of ex-traction methodology on metabolome profiles of Escherichia coli. Anal. Biochem. 2003, 313, 145–154. [Google Scholar] [CrossRef]
- Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T. GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 2005, 579, 1332–1337. [Google Scholar] [CrossRef]
- Fiehn, O.; Barupal, D.K.; Kind, T. Extending biochemical databases by metabolomic surveys. J. Biol Chem. 2011, 286, 23637–23643. [Google Scholar] [CrossRef]
- Khemayan, K.; Pasharawipas, T.; Puiprom, O.; Sriurairatana, S.; Flegel, T.W. Unstable lysogeny and pseudolysogeny in VHS1 bacteriophage of Vibrio harveyi. Appl. Environ. Microbiol. 2006, 72, 1355–1363. [Google Scholar] [CrossRef]
- Zinder, N.D. Lysogenization and superinfection immunity in Salmonella. Virology 1958, 5, 291–326. [Google Scholar] [CrossRef]
- Werts, C.; Michel, V.; Hofnung, M.; Charbit, A. Adsorption of bacteriophage lambda on the LamB protein of Escherichia coli K-12: Point mutations in gene J of lambda responsible for extended host range. J. Bacteriol. 1994, 176, 941–947. [Google Scholar] [CrossRef]
- Kim, M.; Ryu, S. Characterization of a T5-Like Coliphage, SPC35, and Differential Development of Resistance to SPC35 inSalmonella entericaSerovar Typhimurium andEscherichia coli. Appl. Environ. Microbiol. 2011, 77, 2042–2050. [Google Scholar] [CrossRef]
- Ho, T.D.; Slauch, J.M. OmpC is the receptor for Gifsy-1 and Gifsy-2 bacteriophages of Salmonella. J. Bacteriol. 2001, 183, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Piddock, L.J.V. Exploiting the Role of TolC in Pathogenicity: Identification of a Bacteriophage for Eradication of Salmonella Serovars from Poultry. Appl. Environ. Microbiol. 2010, 76, 1704–1706. [Google Scholar] [CrossRef] [PubMed]
- Lenski, R.E.; Levin, B.R. Constraints on the Coevolution of Bacteria and Virulent Phage: A Model, Some Experiments, and Predictions for Natural Communities. Am. Nat. 1985, 125, 585–602. [Google Scholar] [CrossRef]
- Gibson, B.; Wilson, D.J.; Feil, E.; Eyre-Walker, A. The distribution of bacterial doubling times in the wild. Proc. R. Soc. B Biol. Sci. 2018, 285, 20180789. [Google Scholar] [CrossRef]
- León, M.; Kokkari, C.; García, K.; Castillo, D.; Katharios, P.; Bastías, R. Diversification of Vibrio anguillarum Driven by the Bacteriophage CHOED. Front. Microbiol. 2019, 10, 1396. [Google Scholar] [CrossRef]
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. BioEssays 2010, 33, 43–51. [Google Scholar] [CrossRef]
- Pal, C.; Maciá, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nat. Cell Biol. 2007, 450, 1079–1081. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Genet. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Castillo, D.; Christiansen, R.H.; Dalsgaard, I.; Madsen, L.; Middelboe, M. Bacteriophage Resistance Mechanisms in the Fish Pathogen Flavobacterium psychrophilum: Linking Genomic Mutations to Changes in Bacterial Virulence Factors. Appl. Environ. Microbiol. 2014, 81, 1157–1167. [Google Scholar] [CrossRef]
- Bassford, P.J.; Diedrich, D.L.; Schnaitman, C.L.; Reeves, P. Outer membrane proteins of Escherichia coli. VI. Protein alteration in bacteriophage-resistant mutants. J. Bacteriol. 1977, 131, 608–622,. [Google Scholar] [CrossRef]
- Hantke, K. Compilation of Escherichia coli K-12 outer membrane phage receptors–their function and some historical remarks. FEMS Microbiol. Lett. 2020, 367, fnaa013. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Y.; Jiao, N. Phage Resistance of a Marine Bacterium, Roseobacter denitrificans OCh114, as Revealed by Comparative Proteomics. Curr. Microbiol. 2010, 61, 141–147. [Google Scholar] [CrossRef]
- Shin, H.; Lee, J.-H.; Kim, H.; Choi, Y.; Heu, S.; Ryu, S. Receptor Diversity and Host Interaction of Bacteriophages Infecting Salmonella enterica Serovar Typhimurium. PLoS ONE 2012, 7, e43392. [Google Scholar] [CrossRef]
- Gencay, Y.E.; Gambino, M.; Prüssing, T.F.; Brøndsted, L. The genera of bacteriophages and their receptors are the major determinants of host range. Environ. Microbiol. 2019, 21, 2095–2111. [Google Scholar] [CrossRef]
- Yu, F.; Mizushima, S. Roles of lipopolysaccharide and outer membrane protein OmpC of Escherichia coli K-12 in the receptor function for bacteriophage T4. J. Bacteriol. 1982, 151, 718–722. [Google Scholar] [CrossRef]
- Inoue, T.; Matsuzaki, S.; Tanaka, S. A 26-kDa outer membrane protein, OmpK, common to Vibrio species is the receptor for a broad-host-range vibriophage, KVP40. FEMS Microbiol. Lett. 1995, 125, 101–105. [Google Scholar] [CrossRef]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. The Bacteriophage T7 Virion Undergoes Extensive Structural Remodeling during Infection. Science 2013, 339, 576–579. [Google Scholar] [CrossRef]
- Rollauer, S.E.; Sooreshjani, M.A.; Noinaj, N.; Buchanan, S.K. Outer membrane protein biogenesis in Gram-negative bacteria. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150023. [Google Scholar] [CrossRef]
- Laanto, E.; Bamford, J.K.H.; Laakso, J.; Sundberg, L.-R. Phage-Driven Loss of Virulence in a Fish Pathogenic Bacterium. PLoS ONE 2012, 7, e53157. [Google Scholar] [CrossRef]
- Martinez, J.L.; Sánchez, M.B.; Martínez-Solano, L.; Hernandez, A.; Garmendia, L.; Fajardo, A.; Alvarez-Ortega, C. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 2009, 33, 430–449. [Google Scholar] [CrossRef]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Genet. 2013, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Bohannan, B.J.M.; Lenski, R.E. Linking genetic change to community evolution: Insights from studies of bacteria and bacteriophage. Ecol. Lett. 2000, 3, 362–377. [Google Scholar] [CrossRef]
- Chevallereau, A.; Blasdel, B.G.; De Smet, J.; Monot, M.; Zimmermann, M.; Kogadeeva, M.; Sauer, U.; Jorth, P.; Whiteley, M.; Debarbieux, L.; et al. Next-Generation “-omics” Approaches Reveal a Massive Alteration of Host RNA Metabolism during Bacteriophage Infection of Pseudomonas aeruginosa. PLoS Genet. 2016, 12, e1006134. [Google Scholar] [CrossRef] [PubMed]
- Lenski, R.E. Dynamics of interactions between bacteria and virulent bacteriophage. In Advances in Microbial Ecology; Springer: New York, NY, USA, 1988; pp. 1–44. [Google Scholar]
- Brockhurst, M.A.; Rainey, P.B.; Buckling, A. The effect of spatial heterogeneity and parasites on the evolution of host diversity. Proc. R. Soc. B Biol. Sci. 2004, 271, 107–111. [Google Scholar] [CrossRef]
- Heierson, A.; Sidén, I.; Kivaisi, A.; Boman, H.G. Bacteriophage-resistant mutants of Bacillus thuringiensis with decreased virulence in pupae of Hyalophoracecropia. J. Bacteriol. 1986, 167, 18–24. [Google Scholar] [CrossRef]
- Paruchuri, D.K.; Harshey, R.M. Flagellar variation in Serratia marcescens is associated with color variation. J. Bacteriol. 1987, 169, 61–65. [Google Scholar] [CrossRef]
- Brockhurst, M.A.; Buckling, A.; Rainey, P.B. The effect of a bacteriophage on diversification of the opportunistic bacterial pathogen, Pseudomonas aeruginosa. Proc. R. Soc. B Biol. Sci. 2005, 272, 1385–1391. [Google Scholar] [CrossRef]
- Lennon, J.T.; Martiny, J.B.H. Rapid evolution buffers ecosystem impacts of viruses in a microbial food web§. Ecol. Lett. 2008, 11, 1178–1188. [Google Scholar] [CrossRef]
- Middelboe, M.; Holmfeldt, K.; Riemann, L.; Nybroe, O.; Haaber, J.K. Bacteriophages drive strain diversification in a marineFlavobacterium: Implications for phage resistance and physiological properties. Environ. Microbiol. 2009, 11, 1971–1982. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. Nucleotide-dependent Allostery within the ABC Transporter ATP-binding Cassette: A computational study of the mj0796 dimer. J. Biol. Chem. 2007, 282, 22793–22803. [Google Scholar] [CrossRef]
- Mukherjee, S.; Barash, D.; Sengupta, S. Comparative genomics and phylogenomic analyses of lysine riboswitch distributions in bacteria. PLoS ONE 2017, 12, e0184314. [Google Scholar] [CrossRef]
- Juan, C.; Torrens, G.; Barceló, I.M.; Oliver, A. Interplay between Peptidoglycan Biology and Virulence in Gram-Negative Pathogens. Microbiol. Mol. Biol. Rev. 2018, 82, e00033-18. [Google Scholar] [CrossRef]
- Becker, J.; Zelder, O.; Häfner, S.; Schröder, H.; Wittmann, C. From zero to hero—Design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab. Eng. 2011, 13, 159–168. [Google Scholar] [CrossRef]
- Kind, S.; Becker, J.; Wittmann, C. Increased lysine production by flux coupling of the tricarboxylic acid cycle and the lysine biosynthetic pathway metabolic engineering of the availability of succinyl-CoA in Corynebacterium glutamicum. Metab. Eng. 2013, 15, 184–195. [Google Scholar] [CrossRef]
- Xu, J.Z.; Han, M.; Ren, X.D.; Zhang, W.G. Modification of aspartokinase III and dihydrodipicolinate synthetase increases the production of L-lysine in Escherichia coli. Biochem. Eng. J. 2016, 114, 82–89. [Google Scholar] [CrossRef]
- Xu, J.-Z.; Wu, Z.-H.; Gao, S.-J.; Zhang, W. Rational modification of tricarboxylic acid cycle for improving l-lysine production in Corynebacterium glutamicum. Microb. Cell Factories 2018, 17, 105. [Google Scholar] [CrossRef]
- Kamio, Y.; Nakamura, K. Putrescine and cadaverine are constituents of peptidoglycan in Veillonella alcalescens and Veillonella parvula. J. Bacteriol. 1987, 169, 2881–2884. [Google Scholar] [CrossRef]
- Samartzidou, H.; Delcour, A.H. Excretion of Endogenous Cadaverine Leads to a Decrease in Porin-Mediated Outer Membrane Permeability. J. Bacteriol. 1999, 181, 791–798. [Google Scholar] [CrossRef]
- Pereira, C.S.; Santos, A.J.M.; Bejerano-Sagie, M.; Correia, P.B.; Marques, J.C.; Xavier, K.B. Phosphoenolpyruvate phosphotransferase system regulates detection and processing of the quorum sensing signal autoinducer-2. Mol. Microbiol. 2012, 84, 93–104. [Google Scholar] [CrossRef]
- Chatterjee, R.; Millard, C.S.; Champion, K.; Clark, D.P.; Donnelly, M.I. Mutation of the ptsG Gene Results in Increased Production of Succinate in Fermentation of Glucose byEscherichia coli. Appl. Environ. Microbiol. 2001, 67, 148–154. [Google Scholar] [CrossRef]
- Vicente, M.; Chater, K.F.; De Lorenzo, V. Bacterial transcription factors involved in global regulation. Mol. Microbiol. 1999, 33, 8–17. [Google Scholar] [CrossRef]
- Rahman, M.; Hasan, M.R.; Oba, T.; Shimizu, K. Effect of rpoS gene knockout on the metabolism of Escherichia coli during exponential growth phase and early stationary phase based on gene expressions, enzyme activities and intracellular metabolite concentrations. Biotechnol. Bioeng. 2006, 94, 585–595. [Google Scholar] [CrossRef]
- Schellhorn, H.E. Function, Evolution, and Composition of the RpoS Regulon in Escherichia coli. Front. Microbiol. 2020, 11, 2282. [Google Scholar] [CrossRef]
- Zlatkov, N.; Uhlin, B.E. Absence of Global Stress Regulation in Escherichia coli Promotes Pathoadaptation and Novel c-di-GMP-dependent Metabolic Capability. Sci. Rep. 2019, 9, 2600. [Google Scholar] [CrossRef]
- Behr, S.; Fried, L.; Jung, K. Identification of a Novel Nutrient-Sensing Histidine Kinase/Response Regulator Network in Escherichia coli. J. Bacteriol. 2014, 196, 2023–2029. [Google Scholar] [CrossRef]
- Brown, N.L.; Stoyanov, J.V.; Kidd, S.P.; Hobman, J.L. The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 2003, 27, 145–163. [Google Scholar] [CrossRef]
- Thomas, S.A.; Immormino, R.M.; Bourret, R.B.; Silversmith, R.E. Nonconserved Active Site Residues Modulate CheY Autophosphorylation Kinetics and Phosphodonor Preference. Biochemistry 2013, 52, 2262–2273. [Google Scholar] [CrossRef][Green Version]
- del Peso-Santos, T.; Landfors, M.; Skärfstad, E.; Ryden, P.; Shingler, V. Pr is a member of a restricted class of σ70-dependent promoters that lack a recognizable—10 element. Nucleic Acids Res. 2012, 40, 11308–11320. [Google Scholar] [CrossRef]
- Webber, C.A.; Kadner, R.J. Involvement of the amino-terminal phosphorylation module of UhpA in activation of uhpT transcription in Escherichia coli. Mol. Microbiol. 1997, 24, 1039–1048. [Google Scholar] [CrossRef]
- Västermark, Å.; Saier, M.H., Jr. The involvement of transport proteins in transcriptional and metabolic regulation. Curr. Opin. Microbiol. 2014, 18, 8–15. [Google Scholar] [CrossRef]
- Holmfeldt, K.; Solonenko, N.; Shah, M.; Corrier, K.; Riemann, L.; VerBerkmoes, N.C.; Sullivan, M.B. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12798–12803. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Bonnain, C.; Malki, K.; Sawaya, N.A. Phage puppet masters of the marine microbial realm. Nat. Microbiol. 2018, 3, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.M.; Bin Naser, I.; Bari, S.M.N.; Zhu, J.; Mekalanos, J.J.; Faruque, S.M. Quorum Regulated Resistance of Vibrio cholerae against Environmental Bacteriophages. Sci. Rep. 2016, 6, 37956. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.; Nanna, R.; Johanna, J.; Janina, L.; Demeng, T.; Panos, G.K.; Sine, L.S.; Mathias, M. Phage defense mechanisms and their genomic and phenotypic implications in the fish pathogen Vibrio anguillarum. FEMS Microbiol. Ecol. 2019, 95, 3. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Family | Genus | Lifestyle | Genome (bps) | GC % | Predicted # ORFs | Latent Period | Accession Number | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Aphrodite1 | Myoviridae | Aphroditevirus | Lytic | 237.722 | 43.4 | 207 | 80 min | MG720308 | This study |
| phiSt2 | Myoviridae | Schizotequatrovirus | Lytic | 250.485 | 42.6 | 412 | 30 min | KT919973 | [30,31] |
| Ares1 | Siphoviridae | unclassified | Unknown | 80.500 | 45.1 | 119 | 30 min | MG720309 | This study |
| Bacteriophages with High Lytic Activity | |||
|---|---|---|---|
| Vibrio alginolyticus Strains | Aphrodite1 | phiSt2 | Ares1 |
| Control (wild type) | + | + | + |
| VaAphrodite1 | - | + | - |
| VaphiSt2 | + | - | + |
| VaAres1 | + | + | - |
| Gene Name | Nutrient Transport Participation | Type of Transporter | Subunit | Subunit Role |
|---|---|---|---|---|
| mtlA | Mannitol | PTS | IIA, IIB, IIC | Affinity, Transport, Energy |
| treB | Trehalose | PTS | IIB, IIC | Affinity, Transport |
| fruA | Fructose | PTS | IIB, IIC | Affinity, Transport |
| celB | Cellulose | PTS | IIC | Affinity |
| ptsG 1 | Glucose | PTS | IIB, IIC | Affinity, Transport |
| ptsG 2 | Glucose | PTS | IIB, IIC | Affinity, Transport |
| crr | Glucose | PTS | IIB, IIC | Energy |
| ptsN | Nitrogen | PTS | IIA | Transport |
| ptsH | Phosphorus | PTS | IIA | Transport |
| tyrP | Tyrosine | - | - | |
| rbsA | Ribose | ABC | ATP-subunit | Energy |
| metQ | Methionine | ABC | IIA | Affinity |
| metl | Methionine | ABC | Substrate subunit | Transport |
| metN | Methionine | ABC | Transmembrane subunit | Energy |
| artP | Arginine | ABC | Substrate subunit | Affinity |
| artL | Arginine | ABC | ATP-subunit | Energy |
| tcyP | Cysteine | - | - | Transport |
| lysE | Lysine | LysE-like | - | Lysine Export |
| rhtB 1 | Homoserine and Threonine | LysE-like | - | Homoserine and Threonine Export |
| rhtB 2 | Homoserine and Threonine | LysE-like | - | Homoserine and Threonine Export |
| hisP | Lysine, Histidine, Arginine, Ornithine | ABC | ATP-subunit | Energy |
| azlC 1 | Valine, Isoleucine, Leucine | ABC | Transmembrane subunit | Transport |
| azlC 2 | Valine, Isoleucine, Leucine | ABC | Transmembrane subunit | Transport |
| livH | Valine, Isoleucine, Leucine | ABC | Transmembrane subunit | Transport |
| livM | Valine, Isoleucine, Leucine | ABC | Transmembrane subunit | Transport |
| Expression Ratio vs. Control | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | VaAphrodite1 | VaphiSt2 | VaAres1 | |||||||
| livH | * | * | * | |||||||
| livB | * | * | * | |||||||
| metl | * | * | ||||||||
| metN | * | * | ||||||||
| metQ | * | |||||||||
| tcyp | * | * | * | |||||||
| lysE | * | |||||||||
| hisp | * | * | ||||||||
| ptsG1 | * | |||||||||
| ptsG2 | * | * | ||||||||
| crr | * | * | ||||||||
| rbsH | * | * | ||||||||
| cellB | * | * | ||||||||
| ptsH | * | |||||||||
| murE | * | |||||||||
| gltA | * | |||||||||
| frd | * | * | ||||||||
| mdh1 | * | * | ||||||||
| pykA | * | |||||||||
| pykF | ||||||||||
| pckA | * | * | ||||||||
| r > 10 | 4 < r < 10 | 2 < r < 4 | 1.41 < r < 2 | 1.19 < r < 1.41 | 0.84 < r < 1.19 | 0.71 < r < 0.84 | 0.5 < r < 0.71 | 0.25 < r < 0.5 | 0.1 < r < 0.25 | r < 0.1 |
| Gene Name | Coding Enzyme | E.C. Number | Metabolic Pathway |
|---|---|---|---|
| ald | Alanine dehydrogenase | 1.4.11 | Alanine metabolism |
| agxT | Alanine-glycosylate and serine pyruvate aminotransferase | 2.6.1.44 | Alanine metabolism |
| panD | aspartate 4-decarboxylase; desulfinase | 4.1.1.12 | Alanine metabolism |
| lysA 1 | Lysine decarboxylase | 4.1.1.20 | Lysine metabolism |
| lysA 2 | Lysine decarboxylase | 4.1.1.20 | Lysine metabolism |
| murE | Amino acid ligase | 6.3.2.13 | Peptidoglycan biosynthesis |
| gltA | Citrate synthase | 2.3.3.16 | TCA cycle |
| mdh 1 | Malic acid decarboxylase | 1.1.1.37 | TCA cycle |
| frd | Fumarate reductase | 1.3.5.4 | TCA cycle |
| pykA | Pyruvate kinase | 2.7.1.40 | Anaplerotic reactions of the TCA cycle |
| pykF | Pyruvate kinase | 2.7.1.40 | Anaplerotic reactions of the TCA cycle |
| pckA | PEP carboxykinase | 4.1.1.49 | Anaplerotic reactions of the TCA cycle |
| mdh 2 | Malic acid dehydrogenase | 1.1.1.38 | Anaplerotic reactions of the TCA cycle |
| mdh 3 | Malic acid dehydrogenase | 1.1.1.39 | Anaplerotic reactions of the TCA cycle |
| ppc | PEP carboxylase | 4.1.1.31 | Anaplerotic reactions of the TCA cycle |
| Compound | RT | M/Z | Control | VaAphrodite1 | VaphiSt2 | VaAres1 | p |
|---|---|---|---|---|---|---|---|
| Phenylalanine | 25.90 | 218 | 0.046 | 0.068 | 0.079 | 0.087 | 0.093 |
| Lysine | 32.19 | 73 | 0.037 | 0.053 | 0.558 | 0.025 | 0.000 |
| Isoleucine | 17.34 | 158 | 0.074 | 0.091 | 0.113 | 0.105 | 0.129 |
| Alanine | 12.05 | 116 | 0.246 | 0.184 | 0.304 | 0.440 | 0.000 |
| Ornithine | 30.02 | 142 | 0.024 | 0.127 | 0.080 | 0.000 | 0.000 |
| Proline | 17.54 | 142 | 0.059 | 0.000 | 0.057 | 0.072 | 0.000 |
| Valine | 15.19 | 144 | 0.121 | 0.170 | 0.183 | 0.174 | 0.243 |
| Glycine | 17.73 | 174 | 0.084 | 0.157 | 0.121 | 0.219 | 0.032 |
| Leucine | 16.73 | 158 | 0.161 | 0.239 | 0.376 | 0.532 | 0.002 |
| Aspartic acid | 23.21 | 232 | 0.086 | 0.000 | 0.071 | 0.073 | 0.000 |
| Glutamic acid | 25.61 | 246 | 0.248 | 0.116 | 0.486 | 0.343 | 0.000 |
| Pyroglutamic acid | 23.37 | 156 | 0.121 | 0.092 | 0.239 | 0.156 | 0.001 |
| Norleucine | 16.73 | 158 | 0.178 | 0.238 | 0.324 | 0.347 | 0.047 |
| Putrescine | 28.39 | 174 | 0.166 | 0.121 | 0.268 | 0.201 | 0.000 |
| Cadaverine | 30.52 | 174 | 0.717 | 1.120 | 0.361 | 0.507 | 0.000 |
| Succinic acid | 17.92 | 148 | 1.394 | 1.271 | 1.784 | 2.881 | 0.003 |
| Pipecolic acid | 23.37 | 156 | 0.000 | 0.074 | 0.000 | 0.000 | 0.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skliros, D.; Kalatzis, P.G.; Kalloniati, C.; Komaitis, F.; Papathanasiou, S.; Kouri, E.D.; Udvardi, M.K.; Kokkari, C.; Katharios, P.; Flemetakis, E. The Development of Bacteriophage Resistance in Vibrio alginolyticus Depends on a Complex Metabolic Adaptation Strategy. Viruses 2021, 13, 656. https://doi.org/10.3390/v13040656
Skliros D, Kalatzis PG, Kalloniati C, Komaitis F, Papathanasiou S, Kouri ED, Udvardi MK, Kokkari C, Katharios P, Flemetakis E. The Development of Bacteriophage Resistance in Vibrio alginolyticus Depends on a Complex Metabolic Adaptation Strategy. Viruses. 2021; 13(4):656. https://doi.org/10.3390/v13040656
Chicago/Turabian StyleSkliros, Dimitrios, Panos G. Kalatzis, Chrysanthi Kalloniati, Fotios Komaitis, Sokratis Papathanasiou, Evangelia D. Kouri, Michael K. Udvardi, Constantina Kokkari, Pantelis Katharios, and Emmanouil Flemetakis. 2021. "The Development of Bacteriophage Resistance in Vibrio alginolyticus Depends on a Complex Metabolic Adaptation Strategy" Viruses 13, no. 4: 656. https://doi.org/10.3390/v13040656
APA StyleSkliros, D., Kalatzis, P. G., Kalloniati, C., Komaitis, F., Papathanasiou, S., Kouri, E. D., Udvardi, M. K., Kokkari, C., Katharios, P., & Flemetakis, E. (2021). The Development of Bacteriophage Resistance in Vibrio alginolyticus Depends on a Complex Metabolic Adaptation Strategy. Viruses, 13(4), 656. https://doi.org/10.3390/v13040656