
Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination


Abstract
1. Regulation of Innate Immune Signaling by Ubiquitination
2. Ubiquitin as a Versatile Signal to Regulate the Immune Response
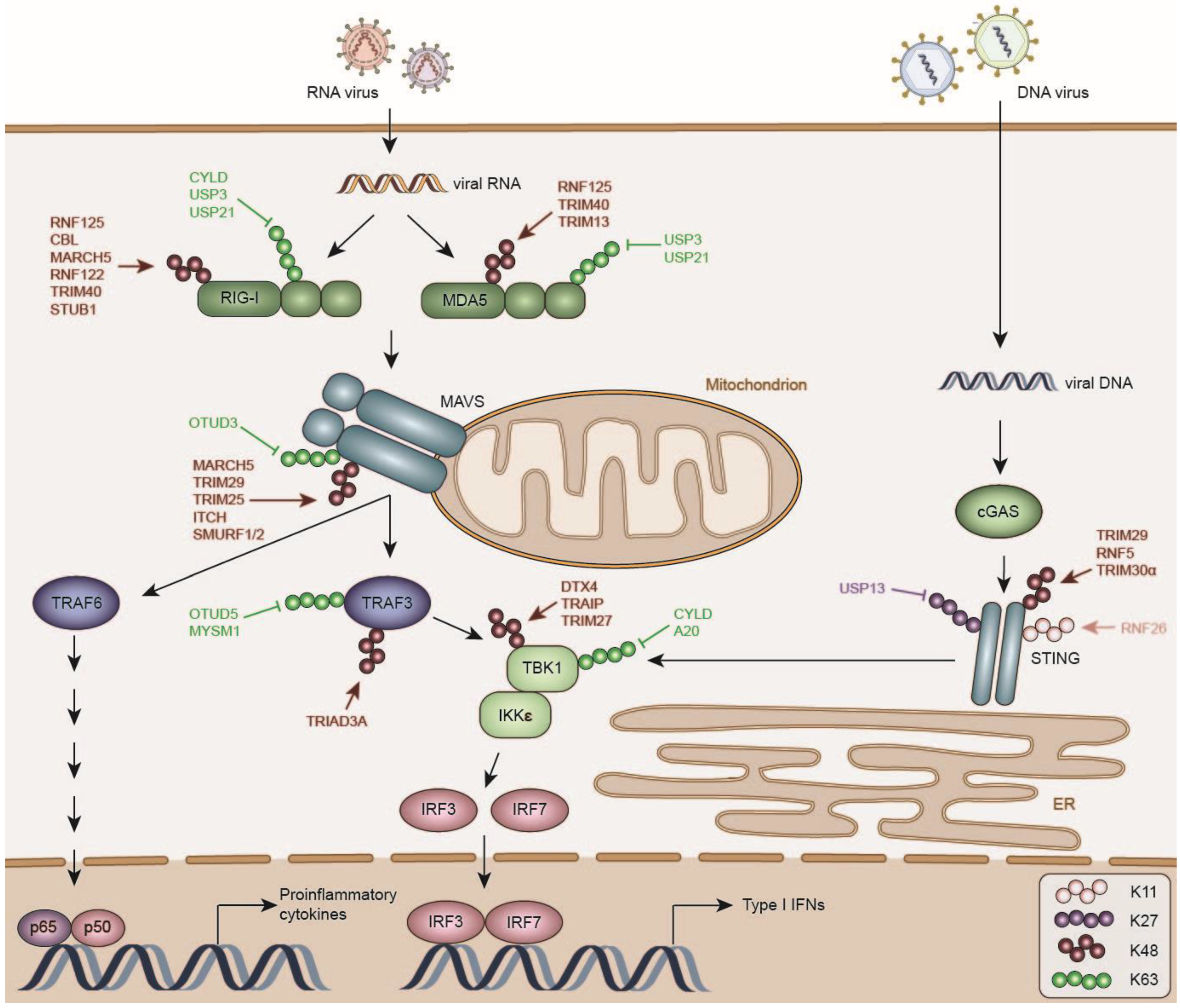
3. Negative Regulation of RLR Signaling
3.1. DUBs Inhibiting RLR Activation
3.2. E3 ligases Destabilizing RLRs
3.2.1. RNF122
3.2.2. TRIM40
3.2.3. TRIM13
3.2.4. RNF125
3.2.5. CBL
3.2.6. STUB1/CHIP
3.2.7. LUBAC
3.3. DUBs Inactivating MAVS
3.4. E3 Ligases Destabilizing MAVS
3.4.1. MARCH5
3.4.2. Itchy E3 Ubiquitin Protein Ligase (ITCH)
3.4.3. TRIM25
3.4.4. TRIM29
3.4.5. Sma and Mothers Against Decapentaplegic Homolog (SMAD) Specific E3 Ubiquitin Protein Ligase (SMURF)1/2
3.5. DUBs Inactivating TBK1
3.6. E3 Ligases Destabilizing TBK1
3.6.1. DTX4
3.6.2. TRAF Interacting Protein (TRAIP)
3.6.3. TRIM27
3.7. DUBs and E3 Ligases Inhibiting TRAF3 Function in Type I IFN Production
3.7.1. OTUD5/Deubiquitinating Enzyme A (DUBA)
3.7.2. MYB-like, Swi3p, Rsc8p and Moira (SWIRM) and MPN Domains 1 (MYSM1)
3.7.3. RNF216
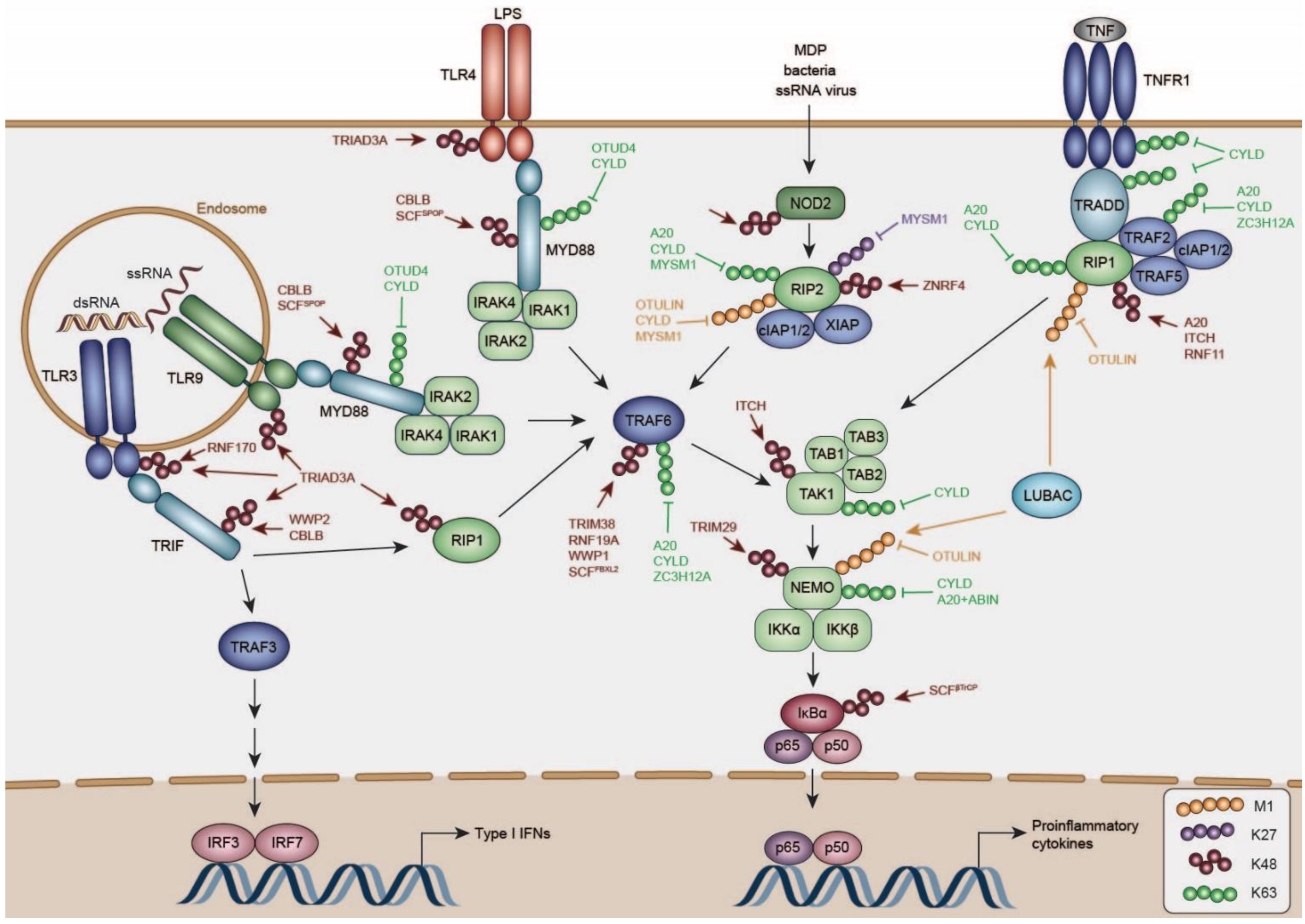
4. Negative Regulation of TLR Signaling
4.1. DUBs Regulating TLR and TLR-Adaptor Activity
DUBs Deactivating MYD88
4.2. E3 Ligases Regulating TLR and TLR-Adaptor Turn-Over
4.2.1. RNF216
4.2.2. RNF170
4.2.3. Casitas B-Lineage Lymphoma Proto-Oncogene B (CBLB)
4.2.4. SCFSPOP
4.2.5. WW Domain Containing E3 Ubiquitin Protein Ligase (WWP)1/2
5. Negative Regulation of Cytosolic DNA Sensor Pathways
DUBs and E3 Ligases Controlling cGAS/STING Activity and Turn-Over
6. Negative Regulation of NLR Signaling
6.1. DUBs Curtailing NOD1/2 Signaling
6.2. Factors Regulating NOD1/2 Protein Levels
7. Negative Regulation of TNF Signaling
7.1. Negative Regulation of RIP1
7.1.1. A20
7.1.2. CYLD
7.1.3. LUBAC and OTULIN
7.2. Negative Regulation of TRAFs in the NFκB Pathway by DUBs and E3 Ligases
7.2.1. CYLD
7.2.2. A20
7.2.3. Zinc Finger CCCH-Type Containing 12A (ZC3H12A)
7.2.4. FBXL2
7.2.5. RNF19A
7.2.6. TRIM38
7.3. Negative Regulation of the TAK1/TAB1/2/3 and IKKα/β/NEMO Complexes
7.3.1. ITCH
7.3.2. TRIM29
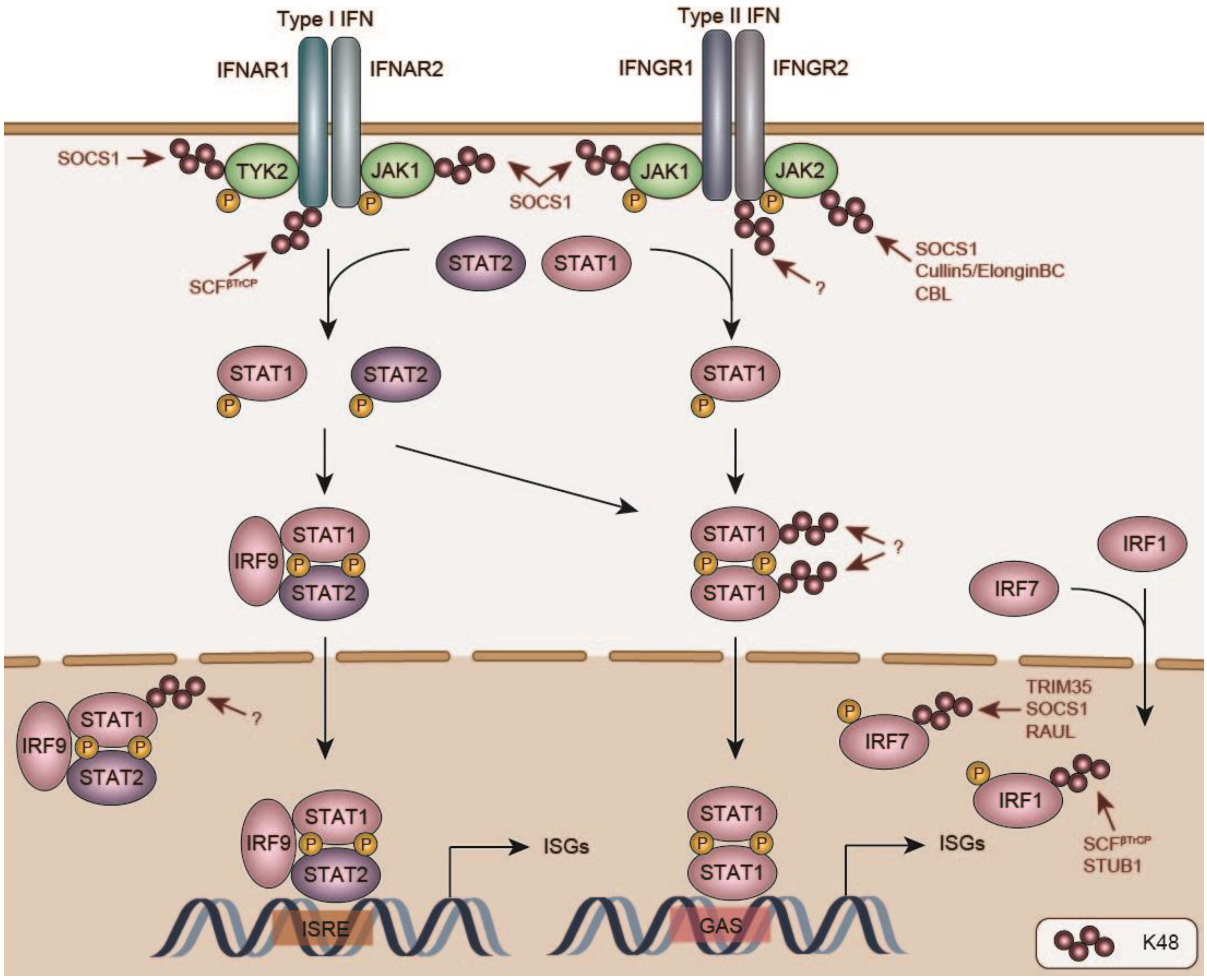
8. Negative Regulation of IFN Signaling and Induced Response Proteins
8.1. IFN Receptors
8.2. Janus Kinases
8.3. STAT Transcription Factors
8.4. Degradation of Innate Immune Response Proteins
9. Concluding Remarks

{kind=link}
{kind=link}
{kind=link}
| Sensor Layer | E3 Ligase | DUB |
|---|---|---|
| RIG-I | RNF125 [74] CBL [75] RNF122 [76] STUB1/CHIP [77,78] TRIM40 [80] MARCH5 [79] | CYLD [67,68] USP14 [71] USP21 [70] USP27X [73] USP3 [69] USP25 [72] |
| MDA5 | RNF125 [74] TRIM40 [80] TRIM13 [84] | USP21 [70] USP3 [69] |
| TRIM25 | LUBAC [12] | |
| cGAS | Unknown [161] | |
| IFI16 | TRIM21 [160] | |
| DDX41 | TRIM21 [159] | |
| NOD2 | TRIM27 [303] RNF34 [304] HSP90/SOCS3 [194] | |
| TLR3, TLR4, TLR5, TLR9 | RNF216/TRIAD3A [123] | |
| TLR3 | RNF170 [139] | |
| TNFR1 | CYLD [189] | |
| IFNAR1 | βTrCP [263,265,305,306] Unknown [307,308,309,310,311,312,313] | |
| IFNGR1 | STUB1/CHIP [314,315] Unknown [266] | |
| Adapter layer | E3 ligase | DUB |
| MAVS | RNF125 [74] SMURF1/2 [106,107] RNF5 [316] ITCH [97,102] MARCH5 [79,96] TRIM25 [103] TRIM29 [105] | OTUD3 [95] YOD1 [317] OTUD1 (indirectly) [108] |
| STING | TRIM29 [104,169] RNF5 [167] TRIM30α [170] RNF26 [168] | USP13 [171] |
| MYD88 | NRDP1 [65] SMURF1 [318] SMURF2 [318] CBLB [140,141] SCFSPOP [142,145,146] | OTUD4 [135] CYLD [137] |
| TRIF | RNF216/TRIAD3A [124] WWP2 [148] CBLB [141] TRIM38 [319,320] | A20 [321] |
| TIRAP | RNF216/TRIAD3A [124] | |
| TRADD | CYLD [189] | |
| TRAF3 | RNF216/TRIAD3A [125] SMURF1 [108,322] PARKIN [323] SCFFBXL2 [236] cIAP1/2 [324] | OTUD5 [119] OTUB1 [325] OTUB2 [325] UCHL1 [326] MYSM1 [122] USP25 [327,328] MCPIP [235] |
| TRAF6 | TRIM38 [240] RNF19A [239] WWP1 [149] CBL [329] RNF11 [216] | USP25 [72] USP2a [330] OTUB1 [325] OTUB2 [325] UCHL1 [326] MYSM1 [122] A20 [216,231,232], (DUB act.-indep.) [233] CYLD [220,221,223,230] MCPIP [235] USP4 [331] |
| TRAF7 | CYLD [230] | |
| TRAF2 | SIAH2 [332] | A20 (DUB act.-indep.) [233] CYLD [220,221,223] MCPIP [235] USP4 [331] |
| cIAP1 | A20 (DUB act.-indep.) [233] | |
| Kinase layer | E3 ligase | DUB |
| RIP1 | RNF216/TRIAD3A [124] A20 [207,211,212,213] ITCH [215] RNF11 [216] | A20 [207,210,211,212] CYLD [189,225,226] OTUD7B [333] USP21 [334] USP4 [335] OTULIN [188] |
| RIP2 | ZNRF4 [195] ITCH [336] (indirectly) | A20 [187] OTULIN [185] CYLD [190] MYSM1 [193] |
| LUBAC (E3 ligase) | OTULIN [188,189,191] | |
| TBK1 | DTX4 [113,114,115] TRAIP [116] TRIM27 [118] SOCS3 [337] TRIM11 [338] RNF144B [339] ASB8 [340] | CYLD [67] USP38 [117] USP2b [341] A20 [110,342] |
| TAK1 | USP18 [343] | |
| TAB2/3 | TRIM30α [344] | |
| NEMO | TRIM29 [244] TRAF7 [345] TRIM40 [346] TRAF4 [347] | CYLD [218,220,223] A20 [210,348] USP18 [343] USP7 [349] UCHL1 [326] OTULIN [191] |
| TAK1 | ITCH [224,242,243] | CYLD [224] USP4 [350] USP19 [351] |
| TAB1 | ITCH [352] | |
| TAB2/3 | HOIL-1 [353] TRIM38 [354] RNF4 [355] | |
| JAK1 | RNF125 [87] Unknown [270,356] | |
| JAK2 | CBL [271,357] SOCS1 [269,358] Cullin5/ElonginBC [269] | USP9X [359] |
| TYK2 | SIAH2 [360] Unknown [361] | |
| Transcription factor layer | E3 ligase | DUB |
| IRF3 | SCF complex [362] HOIL1/RBCK1 [363] TRIM21 [364,365] TRIM22 [364] PIN1+unknown E3 ligase [283] UBE3C/RAUL [8] TRIM26 [366] CBL [367] RNF26 [168] | OTUD1 [368] SENP2 [369] |
| STAT1 | SMURF1 [370] PDLIM2 [371] Unknown [272,274,372,373,374,375] | USP2a [275] USP13 [376] |
| STAT2 | DCST1 [377] FBXW7 [378] | |
| IκBα | SCFβTrCP [379,380] TRIM22 [381] | |
| NFκB | PDLIM2 [382] SOCS1 [383] EloB/C/Cul2/SOCS1 [384,385] MKRN2 [386] RNF182 [387] | |
| Response protein layer | E3 ligase | DUB |
| IRF1 | STUB1/CHIP [278,388] MDM2 [389] βTrCP [300] | |
| IRF7 | SCF complex [288] TRIM21 [390] RAUL/UBE3C [8] SOCS1 [9] SOCS3 [9] NDRG1 [391] TRIM35+PIN1 [290] Unknown [288,299] | |
| RNaseL | Unknown [392,393] | |
| Viperin | UBE4A [394] Unknown [395] | |
| TTP | βTrCP [396] Ub-independent [397] Unknown [398,399,400,401] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Gürtler, C.; Bowie, A.G. Innate Immune Detection of Microbial Nucleic Acids. Trends Microbiol. 2013, 21, 413–420. [Google Scholar] [CrossRef]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the Activation and Attenuation of Innate Antiviral Immunity. J. Exp. Med. 2016, 213, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Gack, M.U. Ubiquitination in the Antiviral Immune Response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin Enzymes in the Regulation of Immune Responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 425–460. [Google Scholar] [CrossRef] [PubMed]
- Kodadek, T.; Sikder, D.; Nalley, K. Keeping Transcriptional Activators under Control. Cell 2006, 127, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hayward, G.S. The Ubiquitin E3 Ligase RAUL Negatively Regulates Type I Interferon through Ubiquitination of the Transcription Factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef]
- Yu, C.-F.; Peng, W.-M.; Schlee, M.; Barchet, W.; Eis-Hübinger, A.M.; Kolanus, W.; Geyer, M.; Schmitt, S.; Steinhagen, F.; Oldenburg, J.; et al. SOCS1 and SOCS3 Target IRF7 Degradation To Suppress TLR7-Mediated Type I IFN Production of Human Plasmacytoid Dendritic Cells. J. Immunol. 2018, 200, 4024–4035. [Google Scholar] [CrossRef]
- Nakagawa, K.; Yokosawa, H. Degradation of Transcription Factor IRF-1 by the Ubiquitin-Proteasome Pathway. The C-Terminal Region Governs the Protein Stability. Eur. J. Biochem. 2000, 267, 1680–1686. [Google Scholar] [CrossRef]
- Diaz-Griffero, F.; Li, X.; Javanbakht, H.; Song, B.; Welikala, S.; Stremlau, M.; Sodroski, J. Rapid Turnover and Polyubiquitylation of the Retroviral Restriction Factor TRIM5. Virology 2006, 349, 300–315. [Google Scholar] [CrossRef]
- Inn, K.-S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.-Y.; Iwai, K.; Jung, J.U. Linear Ubiquitin Assembly Complex Negatively Regulates RIG-I- and TRIM25-Mediated Type I Interferon Induction. Mol. Cell 2011, 41, 354–365. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.; Rape, M. The Increasing Complexity of the Ubiquitin Code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin Chain Diversity at a Glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- van Huizen, M.; Kikkert, M. The Role of Atypical Ubiquitin Chains in the Regulation of the Antiviral Innate Immune Response. Front. Cell Dev. Biol. 2020, 7, 392. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 Enzymes: More than Just Middle Men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like Protein Activation by E1 Enzymes: The Apex for Downstream Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A.P. Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 That Regulates the Organelle’s Dynamics and Signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building Ubiquitin Chains: E2 Enzymes at Work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 Ubiquitin Ligases—Emerging Insights into Their Biological Roles and Disease Relevance. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Walden, H.; Rittinger, K. RBR Ligase–Mediated Ubiquitin Transfer: A Tale with Many Twists and Turns. Nat. Struct. Mol. Biol. 2018, 25, 440–445. [Google Scholar] [CrossRef]
- Dikic, I.; Wakatsuki, S.; Walters, K.J. Ubiquitin-Binding Domains—From Structures to Functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the Chains: Structure and Function of the Deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A Family of Unconventional Deubiquitinases with Modular Chain Specificity Determinants. Nat. Commun. 2018, 9, 799. [Google Scholar] [CrossRef]
- Pinto-Fernández, A.; Davis, S.; Schofield, A.B.; Scott, H.C.; Zhang, P.; Salah, E.; Mathea, S.; Charles, P.D.; Damianou, A.; Bond, G.; et al. Comprehensive Landscape of Active Deubiquitinating Enzymes Profiled by Advanced Chemoproteomics. Front. Chem. 2019, 7, 14. [Google Scholar] [CrossRef]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Malhotra, S.; Morcillo-Suárez, C.; Nurtdinov, R.; Rio, J.; Sarro, E.; Moreno, M.; Castilló, J.; Navarro, A.; Montalban, X.; Comabella, M. Roles of the Ubiquitin Peptidase USP18 in Multiple Sclerosis and the Response to Interferon- β Treatment. Eur. J. Neurol. 2013, 20, 1390–1397. [Google Scholar] [CrossRef]
- Lohr, N.J.; Molleston, J.P.; Strauss, K.A.; Torres-Martinez, W.; Sherman, E.A.; Squires, R.H.; Rider, N.L.; Chikwava, K.R.; Cummings, O.W.; Morton, D.H.; et al. Human ITCH E3 Ubiquitin Ligase Deficiency Causes Syndromic Multisystem Autoimmune Disease. Am. J. Hum. Genet. 2010, 86, 447–453. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.W.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC Deficiency Underlies Autoinflammation, Immunodeficiency, Amylopectinosis, and Lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Priem, D.; van Loo, G.; Bertrand, M.J.M. A20 and Cell Death-Driven Inflammation. Trends Immunol. 2020, 41, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Musone, S.L.; Taylor, K.E.; Lu, T.T.; Nititham, J.; Ferreira, R.C.; Ortmann, W.; Shifrin, N.; Petri, M.A.; Ilyas Kamboh, M.; Manzi, S.; et al. Multiple Polymorphisms in the TNFAIP3 Region Are Independently Associated with Systemic Lupus Erythematosus. Nat. Genet. 2008, 40, 1062–1064. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative Assembly and Dynamic Disassembly of MDA5 Filaments for Viral DsRNA Recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an Adaptor Triggering RIG-I- and Mda5-Mediated Type I Interferon Induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.-M.; Jiang, Z. MAVS Activates TBK1 and IKKε through TRAFs in NEMO Dependent and Independent Manner. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Paz, S.; Vilasco, M.; Werden, S.J.; Arguello, M.; Joseph-Pillai, D.; Zhao, T.; Nguyen, T.L.-A.; Sun, Q.; Meurs, E.F.; Lin, R.; et al. A Functional C-Terminal TRAF3-Binding Site in MAVS Participates in Positive and Negative Regulation of the IFN Antiviral Response. Cell Res. 2011, 21, 895–910. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, Regulation, Modification and Function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-ΚB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347. [Google Scholar] [CrossRef]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural Basis for the Activation of Innate Immune Pattern-Recognition Receptor RIG-I by Viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-Finger E3 Ubiquitin Ligase Is Essential for RIG-I-Mediated Antiviral Activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING Finger Protein, Ubiquitinates RIG-I to Promote Interferon-β Induction during the Early Phase of Viral Infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The Ubiquitin Ligase Riplet Is Essential for RIG-I-Dependent Innate Immune Responses to RNA Virus Infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and Not TRIM25, Is Required for Endogenous RIG-I-Dependent Antiviral Responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.-W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187–1200.e16. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Wu, B.; Xu, H.; Chen, Z.J.; Hur, S. Structural Basis for Ubiquitin-Mediated Antiviral Signal Activation by RIG-I. Nature 2014, 509, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS Forms Functional Prion-Like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The Ubiquitin E3 Ligase TRIM31 Promotes Aggregation and Activation of the Signaling Adaptor MAVS through Lys63-Linked Polyubiquitination. Nat. Immunol. 2017, 18, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural Basis for DsRNA Recognition, Filament Formation, and Antiviral Signal Activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-Catalized Ubiquitination Is Essential for MDA5-Mediated Antiviral Innate Immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef]
- Jiang, X.; Kinch, L.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.; Chen, Z.J. Ubiquitin-Induced Oligomerization of the RNA Sensors RIG-I and MDA5 Activates Antiviral Innate Immune Response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical Role of TRAF3 in the Toll-like Receptor-Dependent and -Independent Antiviral Response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.-P.; Li, S.; Zhong, B.; Li, Y.; Yan, J.; Li, Q.; Teng, C.; Shu, H.-B. Virus-Triggered Ubiquitination of TRAF3/6 by CIAP1/2 Is Essential for Induction of Interferon-β (IFN-β) and Cellular Antiviral Response. J. Biol. Chem. 2010, 285, 9470–9476. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Berman, M.; Kong, Y.-Y.; Dorf, M.E. Mapping a Dynamic Innate Immunity Protein Interaction Network Regulating Type I Interferon Production. Immunity 2011, 35, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Liu, B.; Li, Z.; Wu, H.; Wang, P.; Zhao, K.; Jiang, G.; Zhang, L.; Gao, C. E3 Ubiquitin Ligase RNF128 Promotes Innate Antiviral Immunity through K63-Linked Ubiquitination of TBK1. Nat. Immunol. 2016, 17, 1342–1351. [Google Scholar] [CrossRef]
- Wang, C.; Chen, T.; Zhang, J.; Yang, M.; Li, N.; Xu, X.; Cao, X. The E3 Ubiquitin Ligase Nrdp1 “preferentially” Promotes TLR-Mediated Production of Type I Interferon. Nat. Immunol. 2009, 10, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.Y.; Shen, R.R.; Kim, E.; Lock, Y.J.; Xu, M.; Chen, Z.J.; Hahn, W.C. IKKε-Mediated Tumorigenesis Requires K63-Linked Polyubiquitination by a CIAP1/CIAP2/TRAF2 E3 Ubiquitin Ligase Complex. Cell Rep. 2013, 3, 724–733. [Google Scholar] [CrossRef]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cárdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The Tumour Suppressor CYLD Is a Negative Regulator of RIG-I-Mediated Antiviral Response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, X.; Lee, A.J.; Jin, W.; Chang, M.; Wright, A.; Imaizumi, T.; Sun, S.-C. Regulation of IκB Kinase-Related Kinases and Antiviral Responses by Tumor Suppressor CYLD. J. Biol. Chem. 2008, 283, 18621–18626. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.-F. USP3 Inhibits Type I Interferon Signaling by Deubiquitinating RIG-I-like Receptors. Cell Res. 2014, 24, 400–416. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yu, Y.; Liu, S.; Shi, Z.; Cheng, J.; Zhang, H.; An, L.; Zhao, Y.; Xu, X.; et al. USP21 Negatively Regulates Antiviral Response by Acting as a RIG-I Deubiquitinase. J. Exp. Med. 2014, 211, 313–328. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Ling, J.; Pan, L.; Zhao, X.; Zhu, H.; Yu, J.; Xie, B.; Shen, J.; Chen, W. USP14 Promotes K63-Linked RIG-I Deubiquitination and Suppresses Antiviral Immune Responses. Eur. J. Immunol. 2019, 49, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, D.; Fang, L.; Zhang, H.; Luo, R.; Shang, M.; Ouyang, C.; Ouyang, H.; Chen, H.; Xiao, S. Ubiquitin-Specific Proteases 25 Negatively Regulates Virus-Induced Type I Interferon Signaling. PLoS ONE 2013, 8, e80976. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Chu, B.; Xin, D.; Li, L.; Sun, Q. USP27X Negatively Regulates Antiviral Signaling by Deubiquitinating RIG-I. PLoS Pathog. 2020, 16, e1008293. [Google Scholar] [CrossRef]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative Regulation of the RIG-I Signaling by the Ubiquitin Ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef]
- Chen, W.; Han, C.; Xie, B.; Hu, X.; Yu, Q.; Shi, L.; Wang, Q.; Li, D.; Wang, J.; Zheng, P.; et al. Induction of Siglec-G by RNA Viruses Inhibits the Innate Immune Response by Promoting RIG-I Degradation. Cell 2013, 152, 467–478. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, M.; Liu, S.; Zhang, S.; Liu, W.; Ma, Y.; Zhang, L.; Zhang, J.; Cao, X. RNF122 Suppresses Antiviral Type I Interferon Production by Targeting RIG-I CARDs to Mediate RIG-I Degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 9581–9586. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhang, Q.; Li, X.; Zhao, D.; Liu, Y.; Shen, Q.; Yang, M.; Wang, C.; Li, N.; Cao, X. Cytoplasmic STAT4 Promotes Antiviral Type I IFN Production by Blocking CHIP-Mediated Degradation of RIG-I. J. Immunol. 2016, 196, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ding, X.; Wan, X.; Liu, L.; Yuan, X.; Zhang, W.; Hui, X.; Meng, G.; Xiao, H.; Li, B.; et al. MLL5 Suppresses Antiviral Innate Immune Response by Facilitating STUB1-Mediated RIG-I Degradation. Nat. Commun. 2018, 9, 1243. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Oanh, N.T.K.; Heo, J.; Kim, S.-G.; Lee, H.-S.; Lee, H.; Lee, J.-H.; Kang, H.C.; Lim, W.; Yoo, Y.-S.; et al. Dual Targeting of RIG-I and MAVS by MARCH5 Mitochondria Ubiquitin Ligase in Innate Immunity. Cell. Signal. 2020, 67, 109520. [Google Scholar] [CrossRef]
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623. [Google Scholar] [CrossRef]
- Cui, X.-F.; Imaizumi, T.; Yoshida, H.; Borden, E.C.; Satoh, K. Retinoic Acid-Inducible Gene-I Is Induced by Interferon-Gamma and Regulates the Expression of Interferon-Gamma Stimulated Gene 15 in MCF-7 Cells. Biochem. Cell Biol. Biochim. Biol. Cell. 2004, 82, 401–405. [Google Scholar] [CrossRef]
- Imaizumi, T.; Aratani, S.; Nakajima, T.; Carlson, M.; Matsumiya, T.; Tanji, K.; Ookawa, K.; Yoshida, H.; Tsuchida, S.; McIntyre, T.M.; et al. Retinoic Acid-Inducible Gene-I Is Induced in Endothelial Cells by LPS and Regulates Expression of COX-2. Biochem. Biophys. Res. Commun. 2002, 292, 274–279. [Google Scholar] [CrossRef]
- Zhang, M.; Lee, A.J.; Wu, X.; Sun, S.-C. Regulation of Antiviral Innate Immunity by Deubiquitinase CYLD. Cell. Mol. Immunol. 2011, 8, 502–504. [Google Scholar] [CrossRef][Green Version]
- Narayan, K.; Waggoner, L.; Pham, S.T.; Hendricks, G.L.; Waggoner, S.N.; Conlon, J.; Wang, J.P.; Fitzgerald, K.A.; Kang, J. TRIM13 Is a Negative Regulator of MDA5-Mediated Type I Interferon Production. J. Virol. 2014, 88, 10748–10757. [Google Scholar] [CrossRef]
- Jia, X.; Zhou, H.; Wu, C.; Wu, Q.; Ma, S.; Wei, C.; Cao, Y.; Song, J.; Zhong, H.; Zhou, Z.; et al. The Ubiquitin Ligase RNF125 Targets Innate Immune Adaptor Protein TRIM14 for Ubiquitination and Degradation. J. Immunol. Baltim. 2017, 198, 4652–4658. [Google Scholar] [CrossRef]
- Tang, J.; Tu, S.; Lin, G.; Guo, H.; Yan, C.; Liu, Q.; Huang, L.; Tang, N.; Xiao, Y.; Pope, R.M.; et al. Sequential Ubiquitination of NLRP3 by RNF125 and Cbl-b Limits Inflammasome Activation and Endotoxemia. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Frederick, D.T.; Levesque, M.P.; Cooper, Z.A.; Feng, Y.; Krepler, C.; Brill, L.; Samuels, Y.; Hayward, N.K.; Perlina, A.; et al. Downregulation of the Ubiquitin Ligase RNF125 Underlies Resistance of Melanoma Cells to BRAF Inhibitors via JAK1 Deregulation. Cell Rep. 2015, 11, 1458–1473. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Cao, J.; Zhang, J.-T.; Xu, G.-L.; Li, X.-P.; Wang, F.-T.; Ansari, K.H.; Mohamed, H.; Fan, Y.-Z. Ring Finger Protein 125, as a Potential Highly Aggressive and Unfavorable Prognostic Biomarker, Promotes the Invasion and Metastasis of Human Gallbladder Cancers via Activating the TGF- Β1-SMAD3-ID1 Signaling Pathway. Oncotarget 2017, 8, 49897–49914. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-T.; Chen, L.; Lin, D.S.-C.; Chen, S.-Y.; Tsao, Y.-P.; Guo, H.; Li, F.-J.; Tseng, W.-T.; Tam, J.W.; Chao, C.-W.; et al. NLRP12 Regulates Anti-Viral RIG-I Activation via Interaction with TRIM25. Cell Host Microbe 2019, 25, 602–616.e7. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lai, L.; Chong, Z.; He, J.; Zhang, Y.; Xue, Y.; Xie, Y.; Chen, S.; Dong, P.; Chen, L.; et al. E3 Ligase FBXW7 Is Critical for RIG-I Stabilization during Antiviral Responses. Nat. Commun. 2017, 8, 14654. [Google Scholar] [CrossRef]
- Hoffmann, A.; Kerr, S.; Jellusova, J.; Zhang, J.; Weisel, F.; Wellmann, U.; Winkler, T.H.; Kneitz, B.; Crocker, P.R.; Nitschke, L. Siglec-G Is a B1 Cell-Inhibitory Receptor That Controls Expansion and Calcium Signaling of the B1 Cell Population. Nat. Immunol. 2007, 8, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.; Amanullah, A.; Upadhyay, A.; Mishra, R.; Kumar, A.; Mishra, A. A Decade of Boon or Burden: What Has the CHIP Ever Done for Cellular Protein Quality Control Mechanism Implicated in Neurodegeneration and Aging? Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef]
- Dittmar, G.; Winklhofer, K.F. Linear Ubiquitin Chains: Cellular Functions and Strategies for Detection and Quantification. Front. Chem. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.-K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The Ubiquitin-Specific Protease USP15 Promotes RIG-I-Mediated Antiviral Signaling by Deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fang, X.; Wu, X.; Ling, L.; Chu, F.; Li, J.; Wang, S.; Zang, J.; Zhang, B.; Ye, S.; et al. Acetylation-Dependent Deubiquitinase OTUD3 Controls MAVS Activation in Innate Antiviral Immunity. Mol. Cell 2020, 79, 304–319.e7. [Google Scholar] [CrossRef]
- Yoo, Y.-S.; Park, Y.-Y.; Kim, J.-H.; Cho, H.; Kim, S.-H.; Lee, H.-S.; Kim, T.-H.; Sun Kim, Y.; Lee, Y.; Kim, C.-J.; et al. The Mitochondrial Ubiquitin Ligase MARCH5 Resolves MAVS Aggregates during Antiviral Signalling. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 Mediates Degradation of the Adaptor MAVS via the HECT Ubiquitin Ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; You, F.; Chen, H.; Jiang, Z. Poly(C)-Binding Protein 1 (PCBP1) Mediates Housekeeping Degradation of Mitochondrial Antiviral Signaling (MAVS). Cell Res. 2012, 22, 717–727. [Google Scholar] [CrossRef]
- Allen, I.C.; Moore, C.B.; Schneider, M.; Lei, Y.; Davis, B.K.; Scull, M.A.; Gris, D.; Roney, K.E.; Zimmermann, A.G.; Bowzard, J.B.; et al. NLRX1 Protein Attenuates Inflammatory Responses to Virus Infection by Interfering with the RIG-I-MAVS Signaling Pathway and TRAF6 Ubiquitin Ligase. Immunity 2011, 34, 854–865. [Google Scholar] [CrossRef]
- Qin, Y.; Xue, B.; Liu, C.; Wang, X.; Tian, R.; Xie, Q.; Guo, M.; Li, G.; Yang, D.; Zhu, H. NLRX1 Mediates MAVS Degradation To Attenuate the Hepatitis C Virus-Induced Innate Immune Response through PCBP2. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Rebsamen, M.; Vazquez, J.; Tardivel, A.; Guarda, G.; Curran, J.; Tschopp, J. NLRX1/NOD5 Deficiency Does Not Affect MAVS Signalling. Cell Death Differ. 2011, 18, 1387. [Google Scholar] [CrossRef]
- Choi, Y.B.; Shembade, N.; Parvatiyar, K.; Balachandran, S.; Harhaj, E.W. TAX1BP1 Restrains Virus-Induced Apoptosis by Facilitating Itch-Mediated Degradation of the Mitochondrial Adaptor MAVS. Mol. Cell. Biol. 2016, 37. [Google Scholar] [CrossRef]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidère, N.; Vazquez, A.; Arnoult, D. MAVS Ubiquitination by the E3 Ligase TRIM25 and Degradation by the Proteasome Is Involved in Type I Interferon Production after Activation of the Antiviral RIG-I-like Receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 Promotes DNA Virus Infections by Inhibiting Innate Immune Response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, A.; Minze, L.J.; Li, X.C.; Zhang, Z. TRIM29 Negatively Regulates the Type I IFN Production in Response to RNA Virus. J. Immunol. 2018, 201, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, R.; Meng, J.-L.; Mao, H.-T.; Zhang, Y.; Zhang, J. Smurf2 Negatively Modulates RIG-I–Dependent Antiviral Response by Targeting VISA/MAVS for Ubiquitination and Degradation. J. Immunol. 2014, 192, 4758–4764. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, X.; Ye, X. Ndfip1 Negatively Regulates RIG-I–Dependent Immune Signaling by Enhancing E3 Ligase Smurf1-Mediated MAVS Degradation. J. Immunol. 2012, 189, 5304–5313. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, J.; Qian, L.; Feng, Q.; Wang, X.; Yuan, Y.; Zuo, Y.; Cheng, Q.; Miao, Y.; Guo, T.; et al. Induction of OTUD1 by RNA Viruses Potently Inhibits Innate Immune Responses by Promoting Degradation of the MAVS/TRAF3/TRAF6 Signalosome. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.; Burns, C.; Wicks, I. TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, K.; Barber, G.N.; Harhaj, E.W. TAX1BP1 and A20 Inhibit Antiviral Signaling by Targeting TBK1-IKKi Kinases. J. Biol. Chem. 2010, 285, 14999–15009. [Google Scholar] [CrossRef]
- Iha, H.; Peloponese, J.-M.; Verstrepen, L.; Zapart, G.; Ikeda, F.; Smith, C.D.; Starost, M.F.; Yedavalli, V.; Heyninck, K.; Dikic, I.; et al. Inflammatory Cardiac Valvulitis in TAX1BP1-Deficient Mice through Selective NF-ΚB Activation. EMBO J. 2008, 27, 629–641. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, N.S.; Liebl, D.J.; Harhaj, E.W. Essential Role for TAX1BP1 in the Termination of TNF-Alpha-, IL-1- and LPS-Mediated NF-KappaB and JNK Signaling. EMBO J. 2007, 26, 3910–3922. [Google Scholar] [CrossRef]
- Cui, J.; Li, Y.; Zhu, L.; Liu, D.; Songyang, Z.; Wang, H.Y.; Wang, R.-F. NLRP4 Negatively Regulates Type I Interferon Signaling by Targeting the Kinase TBK1 for Degradation via the Ubiquitin Ligase DTX4. Nat. Immunol. 2012, 13, 387–395. [Google Scholar] [CrossRef] [PubMed]
- An, T.; Li, S.; Pan, W.; Tien, P.; Zhong, B.; Shu, H.-B.; Wu, S. DYRK2 Negatively Regulates Type I Interferon Induction by Promoting TBK1 Degradation via Ser527 Phosphorylation. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef]
- Deng, M.; Tam, J.W.; Wang, L.; Liang, K.; Li, S.; Zhang, L.; Guo, H.; Luo, X.; Zhang, Y.; Petrucelli, A.; et al. TRAF3IP3 Negatively Regulates Cytosolic RNA Induced Anti-Viral Signaling by Promoting TBK1 K48 Ubiquitination. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, L.; Zhao, X.; Zhao, K.; Meng, H.; Zhao, W.; Gao, C. TRAF-Interacting Protein (TRIP) Negatively Regulates IFN-β Production and Antiviral Response by Promoting Proteasomal Degradation of TANK-Binding Kinase 1. J. Exp. Med. 2012, 209, 1703–1711. [Google Scholar] [CrossRef]
- Lin, M.; Zhao, Z.; Yang, Z.; Meng, Q.; Tan, P.; Xie, W.; Qin, Y.; Wang, R.-F.; Cui, J. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination through NLRP4 Signalosome. Mol. Cell 2016, 64, 267–281. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 Suppresses Antiviral Innate Immune Response by Inducing TBK1 Degradation via the Ubiquitin Ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A Deubiquitinase That Regulates Type I Interferon Production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A Histone H2A Deubiquitinase Complex Coordinating Histone Acetylation and H1 Dissociation in Transcriptional Regulation. Mol. Cell 2007, 27, 609–621. [Google Scholar] [CrossRef]
- Fiore, A.; Liang, Y.; Lin, Y.H.; Tung, J.; Wang, H.; Langlais, D.; Nijnik, A. Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review. Int. J. Mol. Sci. 2020, 21, 3007. [Google Scholar] [CrossRef]
- Panda, S.; Nilsson, J.A.; Gekara, N.O. Deubiquitinase MYSM1 Regulates Innate Immunity through Inactivation of TRAF3 and TRAF6 Complexes. Immunity 2015, 43, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-H.; Ulevitch, R.J. Triad3A, an E3 Ubiquitin-Protein Ligase Regulating Toll-like Receptors. Nat. Immunol. 2004, 5, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Fearns, C.; Pan, Q.; Mathison, J.C.; Chuang, T.-H. Triad3A Regulates Ubiquitination and Proteasomal Degradation of RIP1 Following Disruption of Hsp90 Binding. J. Biol. Chem. 2006, 281, 34592–34600. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Mesplede, T.; Solis, M.; Sun, Q.; Zhao, T.; Yang, L.; Chuang, T.-H.; Ware, C.F.; Lin, R.; Hiscott, J. The E3 Ubiquitin Ligase Triad3A Negatively Regulates the RIG-I/MAVS Signaling Pathway by Targeting TRAF3 for Degradation. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Shi, J.-H.; Sun, S.-C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor ΚB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.L.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN Forms a Linear Ubiquitin Ligase Complex Regulating NF-ΚB Activity and Apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of Linear Polyubiquitylation of NEMO in NF-ΚB Activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Xu, M.; Liu, S.; Sun, L.; Chen, Z.J. Key Role of Ubc5 and Lysine-63 Polyubiquitination in Viral Activation of IRF3. Mol. Cell 2009, 36, 315–325. [Google Scholar] [CrossRef]
- Moncrieffe, M.C.; Bollschweiler, D.; Li, B.; Penczek, P.A.; Hopkins, L.; Bryant, C.E.; Klenerman, D.; Gay, N.J. MyD88 Death-Domain Oligomerization Determines Myddosome Architecture: Implications for Toll-like Receptor Signaling. Structure 2020, 28, 281–289.e3. [Google Scholar] [CrossRef]
- Balka, K.R.; De Nardo, D. Understanding Early TLR Signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef]
- Heimdal, K.; Sanchez-Guixé, M.; Aukrust, I.; Bollerslev, J.; Bruland, O.; Jablonski, G.E.; Erichsen, A.K.; Gude, E.; Koht, J.A.; Erdal, S.; et al. STUB1 Mutations in Autosomal Recessive Ataxias—Evidence for Mutation-Specific Clinical Heterogeneity. Orphanet J. Rare Dis. 2014, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mudge, M.C.; Soll, J.M.; Rodrigues, R.B.; Byrum, A.K.; Schwarzkopf, E.A.; Bradstreet, T.R.; Gygi, S.P.; Edelson, B.T.; Mosammaparast, N. OTUD4 Is a Phospho-Activated K63 Deubiquitinase That Regulates MyD88-Dependent Signaling. Mol. Cell 2018, 69, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN Deubiquitinases in NF-ΚB Signaling and Cell Death: So Similar, yet so Different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Miyata, M.; Lim, J.H.; Li, J.-D. Deubiquitinase CYLD Acts as a Negative Regulator for Bacterium NTHi-Induced Inflammation by Suppressing K63-Linked Ubiquitination of MyD88. Proc. Natl. Acad. Sci. USA 2016, 113, E165–E171. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A. Localisation and Trafficking of Toll-like Receptors: An Important Mode of Regulation. Curr. Opin. Immunol. 2010, 22, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, S.; Wang, W.; Ma, Z.; Cao, X.; Jiang, M. E3 Ubiquitin Ligase RNF170 Inhibits Innate Immune Responses by Targeting and Degrading TLR3 in Murine Cells. Cell. Mol. Immunol. 2020, 17, 865–874. [Google Scholar] [CrossRef]
- Bachmaier, K.; Toya, S.; Gao, X.; Triantafillou, T.; Garrean, S.; Park, G.Y.; Frey, R.S.; Vogel, S.; Minshall, R.; Christman, J.W.; et al. E3 Ubiquitin Ligase Cblb Regulates the Acute Inflammatory Response Underlying Lung Injury. Nat. Med. 2007, 13, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Jin, J.; Xu, S.; Liu, H.; Li, N.; Cao, X. Integrin CD11b Negatively Regulates TLR-Triggered Inflammatory Responses by Activating Syk and Promoting Degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 2010, 11, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Guillamot, M.; Ouazia, D.; Dolgalev, I.; Yeung, S.T.; Kourtis, N.; Dai, Y.; Corrigan, K.; Zea-Redondo, L.; Saraf, A.; Florens, L.; et al. The E3 Ubiquitin Ligase SPOP Controls Resolution of Systemic Inflammation by Triggering MYD88 Degradation. Nat. Immunol. 2019, 20, 1196–1207. [Google Scholar] [CrossRef]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-Substrate Complexes: Insights into Molecular Architectures of BTB-Cul3 Ubiquitin Ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef]
- Jiang, J.; Struhl, G. Regulation of the Hedgehog and Wingless Signalling Pathways by the F-Box/WD40-Repeat Protein Slimb. Nature 1998, 391, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, F.; Wang, Q.; Zhang, N.; Zheng, J.; Zheng, M.; Liu, R.; Cui, H.; Wen, J.; Zhao, G. SPOP Promotes Ubiquitination and Degradation of MyD88 to Suppress the Innate Immune Response. PLoS Pathog. 2020, 16, e1008188. [Google Scholar] [CrossRef]
- Jin, X.; Shi, Q.; Li, Q.; Zhou, L.; Wang, J.; Jiang, L.; Zhao, X.; Feng, K.; Lin, T.; Lin, Z.; et al. CRL3–SPOP Ubiquitin Ligase Complex Suppresses the Growth of Diffuse Large B-Cell Lymphoma by Negatively Regulating the MyD88/NF-ΚB Signaling. Leukemia 2020, 34, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 Is an E3 Ubiquitin Ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef]
- Yang, Y.; Liao, B.; Wang, S.; Yan, B.; Jin, Y.; Shu, H.-B.; Wang, Y.-Y. E3 Ligase WWP2 Negatively Regulates TLR3-Mediated Innate Immune Response by Targeting TRIF for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2013, 110, 5115–5120. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-W.; Xu, W.-C.; Luo, J.-G.; Guo, X.-J.; Sun, T.; Zhao, X.-L.; Fu, Z.-J. WW Domain Containing E3 Ubiquitin Protein Ligase 1 (WWP1) Negatively Regulates TLR4-Mediated TNF-α and IL-6 Production by Proteasomal Degradation of TNF Receptor Associated Factor 6 (TRAF6). PLoS ONE 2013, 8, e67633. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type-I Interferon Pathway. Science 2013, 339. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The Helicase DDX41 Senses Intracellular DNA Mediated by the Adaptor STING in Dendritic Cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) Is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Xiao, T.; Fitzgerald, K.A.; et al. IFI16 Is an Innate Immune Sensor for Intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Zheng, J.; Mo, J.; Zhu, T.; Zhuo, W.; Yi, Y.; Hu, S.; Yin, J.; Zhang, W.; Zhou, H.; Liu, Z. Comprehensive Elaboration of the CGAS-STING Signaling Axis in Cancer Development and Immunotherapy. Mol. Cancer 2020, 19. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS–STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Zhang, Z.; Bao, M.; Lu, N.; Weng, L.; Yuan, B.; Liu, Y.-J. The E3 Ubiquitin Ligase TRIM21 Negatively Regulates the Innate Immune Response to Intracellular Double-Stranded DNA. Nat. Immunol. 2013, 14, 172–178. [Google Scholar] [CrossRef]
- Li, D.; Wu, R.; Guo, W.; Xie, L.; Qiao, Z.; Chen, S.; Zhu, J.; Huang, C.; Huang, J.; Chen, B.; et al. STING-Mediated IFI16 Degradation Negatively Controls Type I Interferon Production. Cell Rep. 2019, 29, 1249–1260.e4. [Google Scholar] [CrossRef]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.-F.; et al. TRIM14 Inhibits CGAS Degradation Mediated by Selective Autophagy Receptor P62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-Linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Ni, G.; Konno, H.; Barber, G.N. Ubiquitination of STING at Lysine 224 Controls IRF3 Activation. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, N.; Cui, Y.; Hong, Z.; Liu, X.; Wang, Q.; Li, S.; Liu, H.; Yu, H.; Cai, Y.; et al. The Deubiquitinase CYLD Is a Specific Checkpoint of the STING Antiviral Signaling Pathway. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.-P.; Yang, Y.; Wang, Y.-Y.; Zhang, X.-L.; Shu, H.-B. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhou, M.-T.; Hu, M.-M.; Hu, Y.-H.; Zhang, J.; Guo, L.; Zhong, B.; Shu, H.-B. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PLoS Pathog. 2014, 10, e1004358. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lin, L.; Tong, Y.; Liu, Y.; Mou, J.; Wang, X.; Wang, X.; Gong, Y.; Zhao, Y.; Liu, Y.; et al. TRIM29 Negatively Controls Antiviral Immune Response through Targeting STING for Degradation. Cell Discov. 2018, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lian, Q.; Yang, B.; Yan, S.; Zhou, H.; He, L.; Lin, G.; Lian, Z.; Jiang, Z.; Sun, B. TRIM30α Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PLoS Pathog. 2015, 11, e1005012. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Q.; Jing, Y.-Y.; Zhang, M.; Wang, H.-Y.; Cai, Z.; Liuyu, T.; Zhang, Z.-D.; Xiong, T.-C.; Wu, Y.; et al. USP13 Negatively Regulates Antiviral Responses by Deubiquitinating STING. Nat. Commun. 2017, 8, 15534. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Zheng, C. The Emerging Roles of NOD-like Receptors in Antiviral Innate Immune Signaling Pathways. Int. J. Biol. Macromol. 2021, 169, 407–413. [Google Scholar] [CrossRef]
- Masumoto, J.; Yang, K.; Varambally, S.; Hasegawa, M.; Tomlins, S.A.; Qiu, S.; Fujimoto, Y.; Kawasaki, A.; Foster, S.J.; Horie, Y.; et al. Nod1 Acts as an Intracellular Receptor to Stimulate Chemokine Production and Neutrophil Recruitment in Vivo. J. Exp. Med. 2006, 203, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Chen, F.F.; Muto, A.; Nuñez, G. Human Nod1 Confers Responsiveness to Bacterial Lipopolysaccharides. J. Biol. Chem. 2001, 276, 2551–2554. [Google Scholar] [CrossRef]
- McGovern, D.P.B.; Hysi, P.; Ahmad, T.; van Heel, D.A.; Moffatt, M.F.; Carey, A.; Cookson, W.O.C.; Jewell, D.P. Association between a Complex Insertion/Deletion Polymorphism in NOD1 (CARD4) and Susceptibility to Inflammatory Bowel Disease. Hum. Mol. Genet. 2005, 14, 1245–1250. [Google Scholar] [CrossRef]
- Hugot, J.-P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.-P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 Leucine-Rich Repeat Variants with Susceptibility to Crohn’s Disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A Frameshift Mutation in NOD2 Associated with Susceptibility to Crohn’s Disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Long, Z.; Zhong, F.L.; Teo, D.E.T.; Jin, Y.; Yin, Z.; Boo, Z.Z.; Zhang, Y.; Zhang, J.; Yang, R.; et al. Structural Basis of RIP2 Activation and Signaling. Nat. Commun. 2018, 9, 4993. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, E.; Desfosses, A.; Wallmann, A.; Schulze, W.M.; Rehbein, K.; Mas, P.; Signor, L.; Gaudon, S.; Zenkeviciute, G.; Hons, M.; et al. RIP2 Filament Formation Is Required for NOD2 Dependent NF-ΚB Signalling. Nat. Commun. 2018, 9, 4043. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.M.; Doiron, K.; Labbé, K.; Korneluk, R.G.; Barker, P.A.; Saleh, M. Cellular Inhibitors of Apoptosis CIAP1 and CIAP2 Are Required for Innate Immunity Signaling by the Pattern Recognition Receptors NOD1 and NOD2. Immunity 2009, 30, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.-L.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The Ubiquitin Ligase XIAP Recruits LUBAC for NOD2 Signaling in Inflammation and Innate Immunity. Mol. Cell 2012, 46, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujimoto, Y.; Lucas, P.C.; Nakano, H.; Fukase, K.; Núñez, G.; Inohara, N. A Critical Role of RICK/RIP2 Polyubiquitination in Nod-Induced NF-ΚB Activation. EMBO J. 2008, 27, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN Restricts Met1-Linked Ubiquitination to Control Innate Immune Signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef]
- Abbott, D.W.; Yang, Y.; Hutti, J.E.; Madhavarapu, S.; Kelliher, M.A.; Cantley, L.C. Coordinated Regulation of Toll-Like Receptor and NOD2 Signaling by K63-Linked Polyubiquitin Chains. Mol. Cell. Biol. 2007, 27, 6012–6025. [Google Scholar] [CrossRef]
- Hitotsumatsu, O.; Ahmad, R.-C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The Ubiquitin-Editing Enzyme A20 Restricts Nucleotide-Binding Oligomerization Domain Containing 2-Triggered Signals. Immunity 2008, 28, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN Antagonizes LUBAC Signaling by Specifically Hydrolyzing Met1-Linked Polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef]
- Hrdinka, M.; Fiil, B.K.; Zucca, M.; Leske, D.; Bagola, K.; Yabal, M.; Elliott, P.R.; Damgaard, R.B.; Komander, D.; Jost, P.J.; et al. CYLD Limits Lys63- and Met1-Linked Ubiquitin at Receptor Complexes to Regulate Innate Immune Signaling. Cell Rep. 2016, 14, 2846–2858. [Google Scholar] [CrossRef]
- Takiuchi, T.; Nakagawa, T.; Tamiya, H.; Fujita, H.; Sasaki, Y.; Saeki, Y.; Takeda, H.; Sawasaki, T.; Buchberger, A.; Kimura, T.; et al. Suppression of LUBAC-Mediated Linear Ubiquitination by a Specific Interaction between LUBAC and the Deubiquitinases CYLD and OTULIN. Genes Cells 2014, 19, 254–272. [Google Scholar] [CrossRef]
- Wex, K.; Schmid, U.; Just, S.; Wang, X.; Wurm, R.; Naumann, M.; Schlüter, D.; Nishanth, G. Receptor-Interacting Protein Kinase-2 Inhibition by CYLD Impairs Antibacterial Immune Responses in Macrophages. Front. Immunol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Gekara, N.O. The Deubiquitinase MYSM1 Dampens NOD2-Mediated Inflammation and Tissue Damage by Inactivating the RIP2 Complex. Nat. Commun. 2018, 9, 4654. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Biswas, A.; Liu, Y.-J.; Kobayashi, K.S. Proteasomal Degradation of Nod2 Protein Mediates Tolerance to Bacterial Cell Wall Components. J. Biol. Chem. 2012, 287, 39800–39811. [Google Scholar] [CrossRef] [PubMed]
- Bist, P.; Cheong, W.S.; Ng, A.; Dikshit, N.; Kim, B.-H.; Pulloor, N.K.; Khameneh, H.J.; Hedl, M.; Shenoy, A.R.; Balamuralidhar, V.; et al. E3 Ubiquitin Ligase ZNRF4 Negatively Regulates NOD2 Signalling and Induces Tolerance to MDP. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Williams, K.L.; Lich, J.D.; Duncan, J.A.; Reed, W.; Rallabhandi, P.; Moore, C.; Kurtz, S.; Coffield, V.M.; Accavitti-Loper, M.A.; Su, L.; et al. The CATERPILLER Protein Monarch-1 Is an Antagonist of Toll-like Receptor-, Tumor Necrosis Factor α-, and Mycobacterium Tuberculosis-Induced Pro-Inflammatory Signals. J. Biol. Chem. 2005, 280, 39914–39924. [Google Scholar] [CrossRef] [PubMed]
- Jéru, I.; Duquesnoy, P.; Fernandes-Alnemri, T.; Cochet, E.; Yu, J.W.; Lackmy-Port-Lis, M.; Grimprel, E.; Landman-Parker, J.; Hentgen, V.; Marlin, S.; et al. Mutations in NALP12 Cause Hereditary Periodic Fever Syndromes. Proc. Natl. Acad. Sci. USA 2008, 105, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Griewahn, L.; Köser, A.; Maurer, U. Keeping Cell Death in Check: Ubiquitylation-Dependent Control of TNFR1 and TLR Signaling. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.M.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. CIAP1 and CIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases That Promote RIP1 Ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both CIAP1 and CIAP2 Regulate TNFα-Mediated NF-ΚB Activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific Recognition of Linear Ubiquitin Chains by NEMO Is Important for NF-ΚB Activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.-L.; et al. Linear Ubiquitination Prevents Inflammation and Regulates Immune Signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-ΚB by Ubiquitination and Degradation of the IκBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the Linear Ubiquitin Chain Assembly Complex Stabilizes the TNF-R1 Signaling Complex and Is Required for TNF-Mediated Gene Induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Petersen, S.L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFα Signaling Renders Human Cancer Cells Susceptible to Smac-Mimetic-Induced Apoptosis. Cancer Cell 2007, 12, 445–456. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israël, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, Auto-Inflammation and Amylopectinosis in Humans with Inherited HOIL-1 and LUBAC Deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-Ubiquitination and Ubiquitin Ligase Domains of A20 Downregulate NF-KappaB Signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Krikos, A.; Laherty, C.D.; Dixit, V.M. Transcriptional Activation of the Tumor Necrosis Factor Alpha-Inducible Zinc Finger Protein, A20, Is Mediated by Kappa B Elements. J. Biol. Chem. 1992, 267, 17971–17976. [Google Scholar] [CrossRef]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to Regulate TNF-Induced NF-ΚB and Cell Death Responses in A20-Deficient Mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early Onset Autoinflammatory Syndrome. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef]
- Lu, T.T.; Onizawa, M.; Hammer, G.; Turer, E.E.; Yin, Q.; Damko, E.; Agelides, A.; Shifrin, N.; Advincula, R.; Barrera, J.; et al. Dimerization and Ubiquitin Mediated Recruitment of A20, a Complex Deubiquitinating Enzyme. Immunity 2013, 38, 896–905. [Google Scholar] [CrossRef]
- De, A.; Dainichi, T.; Rathinam, C.V.; Ghosh, S. The Deubiquitinase Activity of A20 Is Dispensable for NF-ΚB Signaling. EMBO Rep. 2014, 15, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Newton, K.; Seshasayee, D.; Kusam, S.; Lam, C.; Zhang, J.; Popovych, N.; Helgason, E.; Schoeffler, A.; Jeet, S.; et al. Phosphorylation and Linear Ubiquitin Direct A20 Inhibition of Inflammation. Nature 2015, 528, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Barford, D. Structure of the A20 OTU Domain and Mechanistic Insights into Deubiquitination. Biochem. J. 2007, 409, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 Ligase Itch Negatively Regulates Inflammatory Signaling Pathways by Controlling the Function of the Ubiquitin-Editing Enzyme A20. Nat. Immunol. 2008, 9, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Parvatiyar, K.; Harhaj, N.S.; Harhaj, E.W. The Ubiquitin-Editing Enzyme A20 Requires RNF11 to Downregulate NF-ΚB Signalling. EMBO J. 2009, 28, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the Familial Cylindromatosis Tumour-Suppressor Gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef]
- Zhang, J.; Stirling, B.; Temmerman, S.T.; Ma, C.A.; Fuss, I.J.; Derry, J.M.J.; Jain, A. Impaired Regulation of NF-ΚB and Increased Susceptibility to Colitis-Associated Tumorigenesis in CYLD-Deficient Mice. J. Clin. Invest. 2006, 116, 3042–3049. [Google Scholar] [CrossRef]
- Massoumi, R.; Chmielarska, K.; Hennecke, K.; Pfeifer, A.; Fässler, R. Cyld Inhibits Tumor Cell Proliferation by Blocking Bcl-3-Dependent NF-ΚB Signaling. Cell 2006, 125, 665–677. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD Is a Deubiquitinating Enzyme That Negatively Regulates NF-KappaB Activation by TNFR Family Members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Nijman, S.M.B.; Dirac, A.M.G.; Bernards, R. Loss of the Cylindromatosis Tumour Suppressor Inhibits Apoptosis by Activating NF-ΚB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Schlicher, L.; Brauns-Schubert, P.; Schubert, F.; Maurer, U. SPATA2: More than a Missing Link. Cell Death Differ. 2017, 24, 1142–1147. [Google Scholar] [CrossRef]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israël, A.; Wallach, D.; Courtois, G. The Tumour Suppressor CYLD Negatively Regulates NF-ΚB Signalling by Deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef]
- Ahmed, N.; Zeng, M.; Sinha, I.; Polin, L.; Wei, W.-Z.; Rathinam, C.; Flavell, R.; Massoumi, R.; Venuprasad, K. The E3 Ligase Itch and Deubiquitinase Cyld Co-Operatively Regulate Tak1 and Inflammation. Nat. Immunol. 2011, 12, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Moquin, D.M.; McQuade, T.; Chan, F.K.-M. CYLD Deubiquitinates RIP1 in the TNFα-Induced Necrosome to Facilitate Kinase Activation and Programmed Necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef]
- Wright, A.; Reiley, W.W.; Chang, M.; Jin, W.; Lee, A.J.; Zhang, M.; Sun, S.-C. Regulation of Early Wave of Germ Cell Apoptosis and Spermatogenesis by Deubiquitinating Enzyme CYLD. Dev. Cell 2007, 13, 705–716. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230.e20. [Google Scholar] [CrossRef] [PubMed]
- Rivkin, E.; Almeida, S.M.; Ceccarelli, D.F.; Juang, Y.-C.; MacLean, T.A.; Srikumar, T.; Huang, H.; Dunham, W.H.; Fukumura, R.; Xie, G.; et al. The Linear Ubiquitin-Specific Deubiquitinase Gumby Regulates Angiogenesis. Nature 2013, 498, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Regamey, A.; Hohl, D.; Liu, J.W.; Roger, T.; Kogerman, P.; Toftgård, R.; Huber, M. The Tumor Suppressor CYLD Interacts with TRIP and Regulates Negatively Nuclear Factor ΚB Activation by Tumor Necrosis Factor. J. Exp. Med. 2003, 198, 1959–1964. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Jono, H.; Kai, H.; Li, J.-D. The Tumor Suppressor Cylindromatosis (CYLD) Acts as a Negative Regulator for Toll-like Receptor 2 Signaling via Negative Cross-Talk with TRAF6 and TRAF7. J. Biol. Chem. 2005, 280, 41111–41121. [Google Scholar] [CrossRef]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.-C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O.; et al. The Ubiquitin-Modifying Enzyme A20 Is Required for Termination of Toll-like Receptor Responses. Nat. Immunol. 2004, 5, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Turer, E.E.; Tavares, R.M.; Mortier, E.; Hitotsumatsu, O.; Advincula, R.; Lee, B.; Shifrin, N.; Malynn, B.A.; Ma, A. Homeostatic MyD88-Dependent Signals Cause Lethal InflamMation in the Absence of A20. J. Exp. Med. 2008, 205, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Ma, A.; Harhaj, E.W. Inhibition of NF-ΚB Signaling by A20 Through Disruption of Ubiquitin Enzyme Complexes. Science 2010, 327, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Yang, R.; Chen, X.; Harhaj, E.W.; Wang, X.; Fan, Y. Regnase-1, a Rapid Response Ribonuclease Regulating Inflammation and Stress Responses. Cell. Mol. Immunol. 2017, 14, 412–422. [Google Scholar] [CrossRef]
- Liang, J.; Saad, Y.; Lei, T.; Wang, J.; Qi, D.; Yang, Q.; Kolattukudy, P.E.; Fu, M. MCP-Induced Protein 1 Deubiquitinates TRAF Proteins and Negatively Regulates JNK and NF-ΚB Signaling. J. Exp. Med. 2010, 207, 2959–2973. [Google Scholar] [CrossRef]
- Chen, B.B.; Coon, T.A.; Glasser, J.R.; McVerry, B.J.; Zhao, J.; Zhao, Y.; Zou, C.; Ellis, B.; Sciurba, F.C.; Zhang, Y.; et al. A Combinatorial F Box Protein Directed Pathway Controls TRAF Adaptor Stability to Regulate Inflammation. Nat. Immunol. 2013, 14, 470–479. [Google Scholar] [CrossRef]
- Mallampalli, R.K.; Coon, T.A.; Glasser, J.R.; Wang, C.; Dunn, S.R.; Weathington, N.M.; Zhao, J.; Zou, C.; Zhao, Y.; Chen, B.B. Targeting F Box Protein Fbxo3 To Control Cytokine-Driven Inflammation. J. Immunol. 2013, 191, 5247–5255. [Google Scholar] [CrossRef]
- Hung, K.-Y.; Liao, W.-I.; Pao, H.-P.; Wu, S.-Y.; Huang, K.-L.; Chu, S.-J. Targeting F-Box Protein Fbxo3 Attenuates Lung Injury Induced by Ischemia-Reperfusion in Rats. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Wu, C.; Su, Z.; Lin, M.; Ou, J.; Zhao, W.; Cui, J.; Wang, R.-F. NLRP11 Attenuates Toll-like Receptor Signalling by Targeting TRAF6 for Degradation via the Ubiquitin Ligase RNF19A. Nat. Commun. 2017, 8, 1977. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Zhang, M.; Yuan, C.; Gao, C. E3 Ubiquitin Ligase Tripartite Motif 38 Negatively Regulates TLR-Mediated Immune Responses by Proteasomal Degradation of TNF Receptor-Associated Factor 6 in Macrophages. J. Immunol. 2012, 188, 2567–2574. [Google Scholar] [CrossRef]
- Matsuda, A.; Suzuki, Y.; Honda, G.; Muramatsu, S.; Matsuzaki, O.; Nagano, Y.; Doi, T.; Shimotohno, K.; Harada, T.; Nishida, E.; et al. Large-Scale Identification and Characterization of Human Genes That Activate NF- κ B and MAPK Signaling Pathways. Oncogene 2003, 22, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Shi, Y.; Liu, S.; Mao, R.; An, L.; Zhao, Y.; Zhang, H.; Zhang, F.; Xu, G.; Qin, J.; et al. Lys48-Linked TAK1 Polyubiquitination at Lysine-72 Downregulates TNFα-Induced NF-ΚB Activation via Mediating TAK1 Degradation. Cell. Signal. 2012, 24, 1381–1389. [Google Scholar] [CrossRef]
- Liang, L.; Fan, Y.; Cheng, J.; Cheng, D.; Zhao, Y.; Cao, B.; Ma, L.; An, L.; Jia, W.; Su, X.; et al. TAK1 Ubiquitination Regulates Doxorubicin-Induced NF-ΚB Activation. Cell. Signal. 2013, 25, 247–254. [Google Scholar] [CrossRef]
- Xing, J.; Weng, L.; Yuan, B.; Wang, Z.; Jia, L.; Jin, R.; Lu, H.; Li, X.C.; Liu, Y.-J.; Zhang, Z. Identification of a Role for TRIM29 in the Control of Innate Immunity in the Respiratory Tract. Nat. Immunol. 2016, 17, 1373–1380. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, Interferon-like Cytokines, and Their Receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Ida-Hosonuma, M.; Iwasaki, T.; Yoshikawa, T.; Nagata, N.; Sato, Y.; Sata, T.; Yoneyama, M.; Fujita, T.; Taya, C.; Yonekawa, H.; et al. The Alpha/Beta Interferon Response Controls Tissue Tropism and Pathogenicity of Poliovirus. J. Virol. 2005, 79, 4460–4469. [Google Scholar] [CrossRef]
- Buchmeier, N.A.; Schreiber, R.D. Requirement of Endogenous Interferon-Gamma Production for Resolution of Listeria Monocytogenes Infection. Proc. Natl. Acad. Sci. USA 1985, 82, 7404–7408. [Google Scholar] [CrossRef] [PubMed]
- Neta, R.; Salvin, S.B. Resistance and Susceptibility to Infection in Inbred Murine Strains. II. Variations in the Effect of Treatment with Thymosin Fraction 5 on the Release of Lymphokines in Vivo. Cell. Immunol. 1983, 75, 173–180. [Google Scholar] [CrossRef]
- Wisseman, C.L.; Waddell, A. Interferonlike Factors from Antigen- and Mitogen-Stimulated Human Leukocytes with Antirickettsial and Cytolytic Actions on Rickettsia Prowazekii. Infected Human Endothelial Cells, Fibroblasts, and Macrophages. J. Exp. Med. 1983, 157, 1780–1793. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Brown, H.M.; Hwang, S. Direct Antiviral Mechanisms of Interferon-Gamma. Immune Netw. 2018, 18, e33. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoité, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.-Y.; Feng, T.; et al. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Kessler, D.S.; Veals, S.A.; Fu, X.Y.; Levy, D.E. Interferon-Alpha Regulates Nuclear Translocation and DNA-Binding Affinity of ISGF3, a Multimeric Transcriptional Activator. Genes Dev. 1990, 4, 1753–1765. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional Regulation by STAT1 and STAT2 in the Interferon JAK-STAT Pathway. JAK-STAT 2013, 2, e23931. [Google Scholar] [CrossRef] [PubMed]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Müller, M.; Novatchkova, M.; Decker, T. A Molecular Switch from STAT2-IRF9 to ISGF3 Underlies Interferon-Induced Gene Transcription. Nat. Commun. 2019, 10, 2921. [Google Scholar] [CrossRef]
- Arimoto, K.-I.; Miyauchi, S.; Stoner, S.A.; Fan, J.-B.; Zhang, D.-E. Negative Regulation of Type I IFN Signaling. J. Leukoc. Biol. 2018. [Google Scholar] [CrossRef]
- Porritt, R.A.; Hertzog, P.J. Dynamic Control of Type I IFN Signalling by an Integrated Network of Negative Regulators. Trends Immunol. 2015, 36, 150–160. [Google Scholar] [CrossRef]
- Higgs, R.; Jefferies, C.A. Targeting IRFs by Ubiquitination: Regulating Antiviral Responses. Biochem. Soc. Trans. 2008, 36, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.S.; Krolewski, J.J.; Fuchs, S.Y. Phosphorylation and Specific Ubiquitin Acceptor Sites Are Required for Ubiquitination and Degradation of the IFNAR1 Subunit of Type I Interferon Receptor. J. Biol. Chem. 2004, 279, 46614–46620. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Kitagawa, M.; Nakayama, K.; Shirane, M.; Matsumoto, M.; Hattori, K.; Higashi, H.; Nakano, H.; Okumura, K.; Onoé, K.; et al. Ubiquitin-Dependent Degradation of IκBα Is Mediated by a Ubiquitin Ligase Skp1/Cul 1/F-Box Protein FWD1. Proc. Natl. Acad. Sci. USA 1999, 96, 3859–3863. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Zheng, H.; HuangFu, W.-C.; Liu, J.; Carbone, C.J.; Leu, N.A.; Baker, D.P.; Fuchs, S.Y. Pathogen Recognition Receptor Signaling Accelerates Phosphorylation-Dependent Degradation of IFNAR1. PLoS Pathog. 2011, 7, e1002065. [Google Scholar] [CrossRef] [PubMed]
- Londino, J.D.; Gulick, D.L.; Lear, T.B.; Suber, T.L.; Weathington, N.M.; Masa, L.S.; Chen, B.B.; Mallampalli, R.K. Post-Translational Modification of the Interferon-Gamma Receptor Alters Its Stability and Signaling. Biochem. J. 2017, 474, 3543–3557. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.S.; Starr, R.; Fenner, J.E.; Scott, C.L.; Handman, E.; Sprigg, N.S.; Corbin, J.E.; Cornish, A.L.; Darwiche, R.; Owczarek, C.M.; et al. SOCS1 Is a Critical Inhibitor of Interferon γ Signaling and Prevents the Potentially Fatal Neonatal Actions of This Cytokine. Cell 1999, 98, 597–608. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Kamizono, S.; Hanada, T.; Yasukawa, H.; Minoguchi, S.; Kato, R.; Minoguchi, M.; Hattori, K.; Hatakeyama, S.; Yada, M.; Morita, S.; et al. The SOCS Box of SOCS-1 Accelerates Ubiquitin-Dependent Proteolysis of TEL-JAK2. J. Biol. Chem. 2001, 276, 12530–12538. [Google Scholar] [CrossRef]
- Lee, Y.; Hyung, S.-W.; Jung, H.J.; Kim, H.-J.; Staerk, J.; Constantinescu, S.N.; Chang, E.-J.; Lee, Z.H.; Lee, S.-W.; Kim, H.-H. The Ubiquitin-Mediated Degradation of Jak1 Modulates Osteoclastogenesis by Limiting Interferon-β–Induced Inhibitory Signaling. Blood 2008, 111, 885–893. [Google Scholar] [CrossRef]
- Lv, K.; Jiang, J.; Donaghy, R.; Riling, C.R.; Cheng, Y.; Chandra, V.; Rozenova, K.; An, W.; Mohapatra, B.C.; Goetz, B.T.; et al. CBL Family E3 Ubiquitin Ligases Control JAK2 Ubiquitination and Stability in Hematopoietic Stem Cells and Myeloid Malignancies. Genes Dev. 2017, 31, 1007–1023. [Google Scholar] [CrossRef]
- Kim, T.K.; Maniatis, T. Regulation of Interferon-Gamma -Activated STAT1 by the Ubiquitin-Proteasome Pathway. Science 1996, 273, 1717–1719. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Yun, H.; Liu, Z.; Su, M.; Lai, R. STAT1β Enhances STAT1 Function by Protecting STAT1α from Degradation in Esophageal Squamous Cell Carcinoma. Cell Death Dis. 2017, 8, e3077. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Liu, Z.; Lai, R. ERK Is a Negative Feedback Regulator for IFN-γ/STAT1 Signaling by Promoting STAT1 Ubiquitination. BMC Cancer 2018, 18, 613. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhao, P.; Liu, J.; Yuan, Y.; Cheng, Q.; Zuo, Y.; Qian, L.; Liu, C.; Guo, T.; Zhang, L.; et al. Deubiquitinase USP2a Sustains Interferons Antiviral Activity by Restricting Ubiquitination of Activated STAT1 in the Nucleus. PLoS Pathog. 2016, 12, e1005764. [Google Scholar] [CrossRef]
- Besche, H.C.; Haas, W.; Gygi, S.P.; Goldberg, A.L. Isolation of Mammalian 26S Proteasomes and P97/VCP Complexes Using the Ubiquitin-like Domain from HHR23B Reveals Novel Proteasome-Associated Proteins. Biochemistry 2009, 48, 2538–2549. [Google Scholar] [CrossRef]
- Narayan, V.; Pion, E.; Landré, V.; Müller, P.; Ball, K.L. Docking-Dependent Ubiquitination of the Interferon Regulatory Factor-1 Tumor Suppressor Protein by the Ubiquitin Ligase CHIP. J. Biol. Chem. 2011, 286, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Eckert, M.; Zylicz, A.; Zylicz, M.; Ball, K.L. Cooperative Regulation of the Interferon Regulatory Factor-1 Tumor Suppressor Protein by Core Components of the Molecular Chaperone Machinery. J. Biol. Chem. 2009, 284, 25889–25899. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; Feng, H.; Rivera-Serrano, E.E.; Selitsky, S.R.; Hirai-Yuki, A.; Das, A.; McKnight, K.L.; Misumi, I.; Hensley, L.; Lovell, W.; et al. Basal Expression of Interferon Regulatory Factor 1 Drives Intrinsic Hepatocyte Resistance to Multiple RNA Viruses. Nat. Microbiol. 2019, 4, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Deng, Y.; Yao, F.; Guan, D.; Feng, Y.; Jiang, H.; Li, X.; Hu, P.; Lu, X.; Wang, H.; et al. Recruitment of Phosphatase PP2A by RACK1 Adaptor Protein Deactivates Transcription Factor IRF3 and Limits Type I Interferon Signaling. Immunity 2014, 40, 515–529. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 Is the Master Regulator of Type-I Interferon-Dependent Immune Responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Visan, I. Deactivating IRF3. Nat. Immunol. 2014, 15, 530. [Google Scholar] [CrossRef]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative Regulation of Interferon-Regulatory Factor 3-Dependent Innate Antiviral Response by the Prolyl Isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef]
- Crosas, B.; Hanna, J.; Kirkpatrick, D.S.; Zhang, D.P.; Tone, Y.; Hathaway, N.A.; Buecker, C.; Leggett, D.S.; Schmidt, M.; King, R.W.; et al. Ubiquitin Chains Are Remodeled at the Proteasome by Opposing Ubiquitin Ligase and Deubiquitinating Activities. Cell 2006, 127, 1401–1413. [Google Scholar] [CrossRef]
- Chu, B.W.; Kovary, K.M.; Guillaume, J.; Chen, L.; Teruel, M.N.; Wandless, T.J. The E3 Ubiquitin Ligase UBE3C Enhances Proteasome Processivity by Ubiquitinating Partially Proteolyzed Substrates. J. Biol. Chem. 2013, 288, 34575–34587. [Google Scholar] [CrossRef]
- Aviram, S.; Kornitzer, D. The Ubiquitin Ligase Hul5 Promotes Proteasomal Processivity. Mol. Cell. Biol. 2010, 30, 985. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFN-α/β Gene Induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Prakash, A.; Levy, D.E. Regulation of IRF7 through Cell Type-Specific Protein Stability. Biochem. Biophys. Res. Commun. 2006, 342, 50–56. [Google Scholar] [CrossRef]
- Maarifi, G.; Smith, N.; Maillet, S.; Moncorgé, O.; Chamontin, C.; Edouard, J.; Sohm, F.; Blanchet, F.P.; Herbeuval, J.-P.; Lutfalla, G.; et al. TRIM8 Is Required for Virus-Induced IFN Response in Human Plasmacytoid Dendritic Cells. Sci. Adv. 2019, 5, eaax3511. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, S.; Yang, B.; Wang, Y.; Zhou, H.; Lian, Q.; Sun, B. TRIM35 Negatively Regulates TLR7- and TLR9-Mediated Type I Interferon Production by Targeting IRF7. FEBS Lett. 2015, 589, 1322–1330. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Type I Interferon-Dependent and -Independent Expression of Tripartite Motif Proteins in Immune Cells. Eur. J. Immunol. 2008, 38, 619–630. [Google Scholar] [CrossRef]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM Gene Expression in Response to Interferons. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Y.-B.; Gui, J.-F.; Lemon, S.M.; Yamane, D. Interferon Regulatory Factor 1 (IRF1) and Anti-Pathogen Innate Immune Responses. PLoS Pathog. 2021, 17, e1009220. [Google Scholar] [CrossRef]
- Chatterjee-Kishore, M.; Wright, K.L.; Ting, J.P.; Stark, G.R. How Stat1 Mediates Constitutive Gene Expression: A Complex of Unphosphorylated Stat1 and IRF1 Supports Transcription of the LMP2 Gene. EMBO J. 2000, 19, 4111–4122. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Stang, M.T.; Liu, Y.; Gao, J.; Ren, B.; Zuckerbraun, B.S.; Mahidhara, R.S.; Xing, Q.; Pizzoferrato, E.; Yim, J.H. Interferon Regulatory Factor 1 (IRF-1) Induces P21(WAF1/CIP1) Dependent Cell Cycle Arrest and P21(WAF1/CIP1) Independent Modulation of Survivin in Cancer Cells. Cancer Lett. 2012, 319, 56–65. [Google Scholar] [CrossRef] [PubMed]
- AbuSara, N.; Razavi, S.; Derwish, L.; Komatsu, Y.; Licursi, M.; Hirasawa, K. Restoration of IRF1-Dependent Anticancer Effects by MEK Inhibition in Human Cancer Cells. Cancer Lett. 2015, 357, 575–581. [Google Scholar] [CrossRef]
- Tanaka, N.; Ishihara, M.; Taniguchi, T. Suppression of C-Myc or FosB-Induced Cell Transformation by the Transcription Factor IRF-1. Cancer Lett. 1994, 83, 191–196. [Google Scholar] [CrossRef]
- Lin, R.; Lin, R.; Hiscott, J.; Hiscott, J.; Hiscott, J. A Role for Casein Kinase II Phosphorylation in the Regulation of IRF-1 Transcriptional Activity. Mol. Cell. Biochem. 1999, 191, 169–180. [Google Scholar] [CrossRef]
- Remoli, A.L.; Sgarbanti, M.; Perrotti, E.; Acchioni, M.; Orsatti, R.; Acchioni, C.; Battistini, A.; Clarke, R.; Marsili, G. IκB Kinase-ε-Mediated Phosphorylation Triggers IRF-1 Degradation in Breast Cancer Cells. Neoplasia 2020, 22, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Garvin, A.J.; Khalaf, A.H.A.; Rettino, A.; Xicluna, J.; Butler, L.; Morris, J.R.; Heery, D.M.; Clarke, N.M. GSK3β-SCFFBXW7α Mediated Phosphorylation and Ubiquitination of IRF1 Are Required for Its Transcription-Dependent Turnover. Nucleic Acids Res. 2019, 47, 4476–4494. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A Diverse Array of Gene Products Are Effectors of the Type I Interferon Antiviral Response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Wolf-Levy, H.; Javitt, A.; Eisenberg-Lerner, A.; Kacen, A.; Ulman, A.; Sheban, D.; Dassa, B.; Fishbain-Yoskovitz, V.; Carmona-Rivera, C.; Kramer, M.P.; et al. Revealing the Cellular Degradome by Mass Spectrometry Analysis of Proteasome-Cleaved Peptides. Nat. Biotechnol. 2018, 36, 1110–1116. [Google Scholar] [CrossRef]
- Zurek, B.; Schoultz, I.; Neerincx, A.; Napolitano, L.M.; Birkner, K.; Bennek, E.; Sellge, G.; Lerm, M.; Meroni, G.; Söderholm, J.D.; et al. TRIM27 Negatively Regulates NOD2 by Ubiquitination and Proteasomal Degradation. PLoS ONE 2012, 7, e41255. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, J.; Song, Y.; Wang, X.; Wang, L.; Xu, J.; Song, C.; Liu, F. The E3 Ligase RNF34 Is a Novel Negative Regulator of the NOD1 Pathway. Cell. Physiol. Biochem. 2014, 33, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; HuangFu, W.-C.; Liu, J.; Veeranki, S.; Baker, D.P.; Koumenis, C.; Diehl, J.A.; Fuchs, S.Y. Inducible Priming Phosphorylation Promotes Ligand-Independent Degradation of the IFNAR1 Chain of Type I Interferon Receptor. J. Biol. Chem. 2010, 285, 2318–2325. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Katlinski, K.V.; Reichert, M.; Takano, S.; Brice, A.; Zhao, B.; Yu, Q.; Zheng, H.; Carbone, C.J.; Katlinskaya, Y.V.; et al. Triggering Ubiquitination of IFNAR1 Protects Tissues from Inflammatory Injury. EMBO Mol. Med. 2014, 6, 384–397. [Google Scholar] [CrossRef]
- Kumar, K.G.S.; Barriere, H.; Carbone, C.J.; Liu, J.; Swaminathan, G.; Xu, P.; Li, Y.; Baker, D.P.; Peng, J.; Lukacs, G.L.; et al. Site-Specific Ubiquitination Exposes a Linear Motif to Promote Interferon-α Receptor Endocytosis. J. Cell Biol. 2007, 179, 935–950. [Google Scholar] [CrossRef]
- Liu, J.; Plotnikov, A.; Banerjee, A.; Suresh Kumar, K.G.; Ragimbeau, J.; Marijanovic, Z.; Baker, D.P.; Pellegrini, S.; Fuchs, S.Y. Ligand-Independent Pathway That Controls Stability of Interferon Alpha Receptor. Biochem. Biophys. Res. Commun. 2008, 367, 388–393. [Google Scholar] [CrossRef][Green Version]
- Zheng, H.; Qian, J.; Varghese, B.; Baker, D.P.; Fuchs, S. Ligand-Stimulated Downregulation of the Alpha Interferon Receptor: Role of Protein Kinase D2. Mol. Cell. Biol. 2011, 31, 710–720. [Google Scholar] [CrossRef]
- Marijanovic, Z.; Ragimbeau, J.; Kumar, K.G.S.; Fuchs, S.Y.; Pellegrini, S. TYK2 Activity Promotes Ligand-Induced IFNAR1 Proteolysis. Biochem. J. 2006, 397, 31–38. [Google Scholar] [CrossRef]
- Zheng, H.; Qian, J.; Baker, D.P.; Fuchs, S.Y. Tyrosine Phosphorylation of Protein Kinase D2 Mediates Ligand-Inducible Elimination of the Type 1 Interferon Receptor. J. Biol. Chem. 2011, 286, 35733–35741. [Google Scholar] [CrossRef]
- Zheng, H.; Qian, J.; Carbone, C.J.; Leu, N.A.; Baker, D.P.; Fuchs, S.Y. Vascular Endothelial Growth Factor-Induced Elimination of the Type 1 Interferon Receptor Is Required for Efficient Angiogenesis. Blood 2011, 118, 4003–4006. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; HuangFu, W.-C.; Kumar, K.G.S.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Virus-Induced Unfolded Protein Response Attenuates Antiviral Defenses via Phosphorylation-Dependent Degradation of the Type I Interferon Receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef]
- Apriamashvili, G.; Vredevoogd, D.W.; Krijgsman, O.; Bleijerveld, O.B.; Ligtenberg, M.A.; de Bruijn, B.; Boshuizen, J.; Altimari, D.D.; Visser, N.L.; Londino, J.D.; et al. Loss of Ubiquitin Ligase STUB1 Amplifies IFNγ-R1/JAK1 Signaling and Sensitizes Tumors to IFNγ. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rebeyev, N. CHIC2 and STUB1 Regulate Interferon-γ Receptor Cell Surface Expression. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2019. [Google Scholar] [CrossRef]
- Zhong, B.; Zhang, Y.; Tan, B.; Liu, T.-T.; Wang, Y.-Y.; Shu, H.-B. The E3 Ubiquitin Ligase RNF5 Targets Virus-Induced Signaling Adaptor for Ubiquitination and Degradation. J. Immunol. 2010, 184, 6249–6255. [Google Scholar] [CrossRef]
- Liu, C.; Huang, S.; Wang, X.; Wen, M.; Zheng, J.; Wang, W.; Fu, Y.; Tian, S.; Li, L.; Li, Z.; et al. The Otubain YOD1 Suppresses Aggregation and Activation of the Signaling Adaptor MAVS through Lys63-Linked Deubiquitination. J. Immunol. 2019, 202, 2957–2970. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.-J.; Park, S.H. Smad6-Specific Recruitment of Smurf E3 Ligases Mediates TGF-Β1-Induced Degradation of MyD88 in TLR4 Signalling. Nat. Commun. 2011, 2, 460. [Google Scholar] [CrossRef]
- Hu, M.-M.; Xie, X.-Q.; Yang, Q.; Liao, C.-Y.; Ye, W.; Lin, H.; Shu, H.-B. TRIM38 Negatively Regulates TLR3/4-Mediated Innate Immune and Inflammatory Responses by Two Sequential and Distinct Mechanisms. J. Immunol. 2015, 195, 4415–4425. [Google Scholar] [CrossRef]
- Xue, Q.; Zhou, Z.; Lei, X.; Liu, X.; He, B.; Wang, J.; Hung, T. TRIM38 Negatively Regulates TLR3-Mediated IFN-β Signaling by Targeting TRIF for Degradation. PLoS ONE 2012, 7, e46825. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Li, L.; Han, K.-J.; Zhai, Z.; Shu, H.-B. A20 Is a Potent Inhibitor of TLR3- and Sendai Virus-Induced Activation of NF-ΚB and ISRE and IFN-β Promoter. FEBS Lett. 2004, 576, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, K.; Wang, J.; An, L.; Yang, G.; Chen, H.; Cui, Y.; Yin, X.; Xie, P.; Xing, G.; et al. Ubiquitin Ligase Smurf1 Targets TRAF Family Proteins for Ubiquitination and Degradation. Mol. Cell. Biochem. 2010, 338, 11–17. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, R.; Zhang, Z.; Liu, Q.; Zhang, Y.; Wang, Q.; Yuan, H. Mitochondrial Protein PINK1 Positively Regulates RLR Signaling. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Tseng, P.-H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.A.; Karin, M. Different Modes of TRAF3 Ubiquitination Selectively Activate Type I Interferon and Proinflammatory Cytokine Expression. Nat. Immunol. 2010, 11, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zheng, H.; Mao, A.-P.; Zhong, B.; Li, Y.; Liu, Y.; Gao, Y.; Ran, Y.; Tien, P.; Shu, H.-B. Regulation of Virus-Triggered Signaling by OTUB1- and OTUB2-Mediated Deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 2010, 285, 4291–4297. [Google Scholar] [CrossRef]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J.M.; et al. Human Papillomavirus (HPV) Upregulates the Cellular Deubiquitinase UCHL1 to Suppress the Keratinocyte’s Innate Immune Response. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Zhong, B.; Liu, X.; Wang, X.; Liu, X.; Li, H.; Darnay, B.G.; Lin, X.; Sun, S.-C.; Dong, C. Ubiquitin-Specific Protease 25 Regulates TLR4-Dependent Innate Immune Responses Through Deubiquitination of the Adaptor Protein TRAF3. Sci. Signal. 2013, 6, ra35. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, M.; Zhang, M.-X.; Ren, Y.; Jin, J.; Zhao, Q.; Pan, Z.; Wu, M.; Shu, H.-B.; Dong, C.; et al. Induction of USP25 by Viral Infection Promotes Innate Antiviral Responses by Mediating the Stabilization of TRAF3 and TRAF6. Proc. Natl. Acad. Sci. USA 2015, 112, 11324–11329. [Google Scholar] [CrossRef]
- Jang, H.-D.; Hwang, H.Z.; Kim, H.-S.; Lee, S.Y. C-Cbl Negatively Regulates TRAF6-Mediated NF-ΚB Activation by Promoting K48-Linked Polyubiquitination of TRAF6. Cell. Mol. Biol. Lett. 2019, 24. [Google Scholar] [CrossRef]
- He, X.; Li, Y.; Li, C.; Liu, L.-J.; Zhang, X.-D.; Liu, Y.; Shu, H.-B. USP2a Negatively Regulates IL-1β- and Virus-Induced NF-ΚB Activation by Deubiquitinating TRAF6. J. Mol. Cell Biol. 2013, 5, 39–47. [Google Scholar] [CrossRef]
- Xiao, N.; Li, H.; Luo, J.; Wang, R.; Chen, H.; Chen, J.; Wang, P. Ubiquitin-Specific Protease 4 (USP4) Targets TRAF2 and TRAF6 for Deubiquitination and Inhibits TNFα-Induced Cancer Cell Migration. Biochem. J. 2012, 441, 979–986. [Google Scholar] [CrossRef]
- Habelhah, H.; Frew, I.J.; Laine, A.; Janes, P.W.; Relaix, F.; Sassoon, D.; Bowtell, D.D.L.; Ronai, Z. Stress-Induced Decrease in TRAF2 Stability Is Mediated by Siah2. EMBO J. 2002, 21, 5756–5765. [Google Scholar] [CrossRef] [PubMed]
- Enesa, K.; Zakkar, M.; Chaudhury, H.; Luong, L.A.; Rawlinson, L.; Mason, J.C.; Haskard, D.O.; Dean, J.L.E.; Evans, P.C. NF-KappaB Suppression by the Deubiquitinating Enzyme Cezanne: A Novel Negative Feedback Loop in pro-Inflammatory Signaling. J. Biol. Chem. 2008, 283, 7036–7045. [Google Scholar] [CrossRef]
- Xu, G.; Tan, X.; Wang, H.; Sun, W.; Shi, Y.; Burlingame, S.; Gu, X.; Cao, G.; Zhang, T.; Qin, J.; et al. Ubiquitin-Specific Peptidase 21 Inhibits Tumor Necrosis Factor α-Induced Nuclear Factor ΚB Activation via Binding to and Deubiquitinating Receptor-Interacting Protein 1. J. Biol. Chem. 2010, 285, 969–978. [Google Scholar] [CrossRef]
- Hou, X.; Wang, L.; Zhang, L.; Pan, X.; Zhao, W. Ubiquitin-Specific Protease 4 Promotes TNF-α-Induced Apoptosis by Deubiquitination of RIP1 in Head and Neck Squamous Cell Carcinoma. FEBS Lett. 2013, 587, 311–316. [Google Scholar] [CrossRef]
- Tigno-Aranjuez, J.T.; Bai, X.; Abbott, D.W. A Discrete Ubiquitin-Mediated Network Regulates the Strength of NOD2 Signaling. Mol. Cell. Biol. 2013, 33, 146–158. [Google Scholar] [CrossRef][Green Version]
- Liu, D.; Sheng, C.; Gao, S.; Yao, C.; Li, J.; Jiang, W.; Chen, H.; Wu, J.; Pan, C.; Chen, S.; et al. SOCS3 Drives Proteasomal Degradation of TBK1 and Negatively Regulates Antiviral Innate Immunity. Mol. Cell. Biol. 2015, 35, 2400–2413. [Google Scholar] [CrossRef]
- Lee, Y.; Song, B.; Park, C.; Kwon, K.-S. TRIM11 Negatively Regulates IFNβ Production and Antiviral Activity by Targeting TBK1. PLoS ONE 2013, 8, e63255. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Wang, B.; Zhu, X.; Zhao, L.; Chu, C.; Guo, Q.; Wei, R.; Yin, X.; Zhang, Y.; et al. RNF144B Inhibits LPS-Induced Inflammatory Responses via Binding TBK1. J. Leukoc. Biol. 2019, 106, 1303–1311. [Google Scholar] [CrossRef]
- Guo, Y.; Li, R.; Tan, Z.; Shi, J.; Fu, Y.; Song, Y.; Zhu, M.; Zhang, L.; Huang, J. E3 Ubiquitin Ligase ASB8 Negatively Regulates Interferon via Regulating TBK1/IKKi Homeostasis. Mol. Immunol. 2020, 121, 195–203. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, X.; Zhang, M.; Zhao, W.; Gao, C. Ubiquitin-Specific Protease 2b Negatively Regulates IFN-β Production and Antiviral Activity by Targeting TANK-Binding Kinase 1. J. Immunol. 2014, 193, 2230–2237. [Google Scholar] [CrossRef]
- Mérour, E.; Jami, R.; Lamoureux, A.; Bernard, J.; Brémont, M.; Biacchesi, S. A20 (Tnfaip3) Is a Negative Feedback Regulator of RIG-I-Mediated IFN Induction in Teleost. Fish. Shellfish Immunol. 2019, 84, 857–864. [Google Scholar] [CrossRef]
- Yang, Z.; Xian, H.; Hu, J.; Tian, S.; Qin, Y.; Wang, R.-F.; Cui, J. USP18 Negatively Regulates NF-ΚB Signaling by Targeting TAK1 and NEMO for Deubiquitination through Distinct Mechanisms. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. TRIM30α Negatively Regulates TLR-Mediated NF-ΚB Activation by Targeting TAB2 and TAB3 for Degradation. Nat. Immunol. 2008, 9, 369–377. [Google Scholar] [CrossRef]
- Zotti, T.; Uva, A.; Ferravante, A.; Vessichelli, M.; Scudiero, I.; Ceccarelli, M.; Vito, P.; Stilo, R. TRAF7 Protein Promotes Lys-29-Linked Polyubiquitination of IκB Kinase (IKKγ)/NF-ΚB Essential Modulator (NEMO) and P65/RelA Protein and Represses NF-ΚB Activation. J. Biol. Chem. 2011, 286, 22924–22933. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Okumura, F.; Takahashi, N.; Kataoka, A.; Kamiyama, T.; Todo, S.; Hatakeyama, S. TRIM40 Promotes Neddylation of IKKγ and Is Downregulated in Gastrointestinal Cancers. Carcinogenesis 2011, 32, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Marinis, J.M.; Homer, C.R.; McDonald, C.; Abbott, D.W. A Novel Motif in the Crohn’s Disease Susceptibility Protein, NOD2, Allows TRAF4 to Down-Regulate Innate Immune Responses. J. Biol. Chem. 2011, 286, 1938–1950. [Google Scholar] [CrossRef]
- Mauro, C.; Pacifico, F.; Lavorgna, A.; Mellone, S.; Iannetti, A.; Acquaviva, R.; Formisano, S.; Vito, P.; Leonardi, A. ABIN-1 Binds to NEMO/IKKγ and Co-Operates with A20 in Inhibiting NF-ΚB*. J. Biol. Chem. 2006, 281, 18482–18488. [Google Scholar] [CrossRef]
- Li, T.; Guan, J.; Li, S.; Zhang, X.; Zheng, X. HSCARG Downregulates NF-ΚB Signaling by Interacting with USP7 and Inhibiting NEMO Ubiquitination. Cell Death Dis. 2014, 5, e1229. [Google Scholar] [CrossRef]
- Fan, Y.-H.; Yu, Y.; Mao, R.-F.; Tan, X.-J.; Xu, G.-F.; Zhang, H.; Lu, X.-B.; Fu, S.-B.; Yang, J. USP4 Targets TAK1 to Downregulate TNF α -Induced NF- κ B Activation. Cell Death Differ. 2011, 18, 1547–1560. [Google Scholar] [CrossRef]
- Lei, C.-Q.; Wu, X.; Zhong, X.; Jiang, L.; Zhong, B.; Shu, H.-B. USP19 Inhibits TNF-α– and IL-1β–Triggered NF-ΚB Activation by Deubiquitinating TAK1. J. Immunol. 2019, 203, 259–268. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Kathania, M.; Zeng, M.; Anguiano, E.; Basrur, V.; Vandergriff, T.; Pascual, V.; Wei, W.-Z.; Massoumi, R.; Venuprasad, K. The E3 Ubiquitin Ligase Itch Inhibits P38α Signaling and Skin Inflammation through the Ubiquitylation of Tab1. Sci. Signal. 2015, 8, ra22. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, Y.; Zhong, B.; Wang, Y.-Y.; Diao, F.-C.; Wang, R.-P.; Zhang, M.; Chen, D.-Y.; Zhai, Z.-H.; Shu, H.-B. RBCK1 Negatively Regulates Tumor Necrosis Factor- and Interleukin-1-Triggered NF-ΚB Activation by Targeting TAB2/3 for Degradation. J. Biol. Chem. 2007, 282, 16776–16782. [Google Scholar] [CrossRef]
- Hu, M.-M.; Yang, Q.; Zhang, J.; Liu, S.-M.; Zhang, Y.; Lin, H.; Huang, Z.-F.; Wang, Y.-Y.; Zhang, X.-D.; Zhong, B.; et al. TRIM38 Inhibits TNFα- and IL-1β–Triggered NF-ΚB Activation by Mediating Lysosome-Dependent Degradation of TAB2/3. Proc. Natl. Acad. Sci. USA 2014, 111, 1509–1514. [Google Scholar] [CrossRef]
- Tan, B.; Mu, R.; Chang, Y.; Wang, Y.-B.; Wu, M.; Tu, H.-Q.; Zhang, Y.-C.; Guo, S.-S.; Qin, X.-H.; Li, T.; et al. RNF4 Negatively Regulates NF-ΚB Signaling by down-Regulating TAB2. FEBS Lett. 2015, 589, 2850–2858. [Google Scholar] [CrossRef]
- Yu, C.-L.; Burakoff, S.J. Involvement of Proteasomes in Regulating Jak-STAT Pathways upon Interleukin-2 Stimulation. J. Biol. Chem. 1997, 272, 14017–14020. [Google Scholar] [CrossRef]
- Nagao, T.; Oshikawa, G.; Wu, N.; Kurosu, T.; Miura, O. DNA Damage Stress and Inhibition of Jak2-V617F Cause Its Degradation and Synergistically Induce Apoptosis through Activation of GSK3β. PLoS ONE 2011, 6, e27397. [Google Scholar] [CrossRef]
- Ungureanu, D.; Saharinen, P.; Junttila, I.; Hilton, D.J.; Silvennoinen, O. Regulation of Jak2 through the Ubiquitin-Proteasome Pathway Involves Phosphorylation of Jak2 on Y1007 and Interaction with SOCS-1. Mol. Cell. Biol. 2002, 22, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Umezawa, Y.; Watanabe, D.; Okada, K.; Ishida, S.; Nogami, A.; Miura, O. Inhibition of USP9X Downregulates JAK2-V617F and Induces Apoptosis Synergistically with BH3 Mimetics Preferentially in Ruxolitinib-Persistent JAK2-V617F-Positive Leukemic Cells. Cancers 2020, 12, 406. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Chen, Y.; Ginter, T.; Schäfer, C.; Buchwald, M.; Schmitz, M.L.; Klitzsch, J.; Schütz, A.; Haitel, A.; Schmid, K.; et al. SIAH2 Antagonizes TYK2-STAT3 Signaling in Lung Carcinoma Cells. Oncotarget 2014, 5, 3184–3196. [Google Scholar] [CrossRef]
- Akahane, K.; Sanda, T.; Mansour, M.R.; Radimerski, T.; DeAngelo, D.J.; Weinstock, D.M.; Look, A.T. HSP90 Inhibition Leads to Degradation of the TYK2 Kinase and Apoptotic Cell Death in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2016, 30, 219–228. [Google Scholar] [CrossRef]
- Bibeau-Poirier, A.; Gravel, S.-P.; Clément, J.-F.; Rolland, S.; Rodier, G.; Coulombe, P.; Hiscott, J.; Grandvaux, N.; Meloche, S.; Servant, M.J. Involvement of the IκB Kinase (IKK)-Related Kinases Tank-Binding Kinase 1/IKKi and Cullin-Based Ubiquitin Ligases in IFN Regulatory Factor-3 Degradation. J. Immunol. 2006, 177, 5059–5067. [Google Scholar] [CrossRef]
- Zhang, M.; Tian, Y.; Wang, R.-P.; Gao, D.; Zhang, Y.; Diao, F.-C.; Chen, D.-Y.; Zhai, Z.-H.; Shu, H.-B. Negative Feedback Regulation of Cellular Antiviral Signaling by RBCK1-Mediated Degradation of IRF3. Cell Res. 2008, 18, 1096–1104. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, W.; Fan, W.; Wu, Y.; Zhang, M.; Xu, J.; Li, W.; Sun, L.; Liu, W.; Liu, W. Forkhead Box O1-Mediated Ubiquitination Suppresses RIG-I-Mediated Antiviral Immune Responses. Int. Immunopharmacol. 2021, 90, 107152. [Google Scholar] [CrossRef]
- Higgs, R.; Ní Gabhann, J.; Ben Larbi, N.; Breen, E.P.; Fitzgerald, K.A.; Jefferies, C.A. The E3 Ubiquitin Ligase Ro52 Negatively Regulates IFN-Beta Production Post-Pathogen Recognition by Polyubiquitin-Mediated Degradation of IRF3. J. Immunol. Baltim. Md 1950 2008, 181, 1780–1786. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 Negatively Regulates Interferon-β Production and Antiviral Response through Polyubiquitination and Degradation of Nuclear IRF3. PLOS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, H.; Yu, J.; Li, H.; Ge, J.; Chen, W. C-Cbl-Mediated Ubiquitination of IRF3 Negatively Regulates IFN-β Production and Cellular Antiviral Response. Cell. Signal. 2016, 28, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, D.; Wang, P.; Zhao, Y.; You, F. OTUD1 Negatively Regulates Type I IFN Induction by Disrupting Noncanonical Ubiquitination of IRF3. J. Immunol. 2020, 204, 1904–1918. [Google Scholar] [CrossRef]
- Ran, Y.; Liu, T.-T.; Zhou, Q.; Li, S.; Mao, A.-P.; Li, Y.; Liu, L.-J.; Cheng, J.-K.; Shu, H.-B. SENP2 Negatively Regulates Cellular Antiviral Response by DeSUMOylating IRF3 and Conditioning It for Ubiquitination and Degradation. J. Mol. Cell Biol. 2011, 3, 283–292. [Google Scholar] [CrossRef]
- Yuan, C.; Qi, J.; Zhao, X.; Gao, C. Smurf1 Protein Negatively Regulates Interferon-γ Signaling through Promoting STAT1 Protein Ubiquitination and Degradation. J. Biol. Chem. 2012, 287, 17006–17015. [Google Scholar] [CrossRef]
- Tanaka, T.; Soriano, M.A.; Grusby, M.J. SLIM Is a Nuclear Ubiquitin E3 Ligase That Negatively Regulates STAT Signaling. Immunity 2005, 22, 729–736. [Google Scholar] [CrossRef]
- Gao, C.; Guo, H.; Mi, Z.; Kuo, P.C. Osteopontin Induces Ubiquitin-Dependent Degradation of STAT1 in RAW264.7 Murine Macrophages. J. Immunol. 2007, 178, 1870–1881. [Google Scholar] [CrossRef]
- Yu, N.; Xue, M.; Wang, W.; Xia, D.; Li, Y.; Zhou, X.; Pang, D.; Lu, K.; Hou, J.; Zhang, A.; et al. RNF168 Facilitates Proliferation and Invasion of Esophageal Carcinoma, Possibly via Stabilizing STAT1. J. Cell. Mol. Med. 2019, 23, 1553–1561. [Google Scholar] [CrossRef]
- Haspel, R.L.; Salditt-Georgieff, M.; Darnell, J.E. The Rapid Inactivation of Nuclear Tyrosine Phosphorylated Stat1 Depends upon a Protein Tyrosine Phosphatase. EMBO J. 1996, 15, 6262–6268. [Google Scholar] [CrossRef]
- Zuo, Y.; Feng, Q.; Jin, L.; Huang, F.; Miao, Y.; Liu, J.; Xu, Y.; Chen, X.; Zhang, H.; Guo, T.; et al. Regulation of the Linear Ubiquitination of STAT1 Controls Antiviral Interferon Signaling. Nat. Commun. 2020, 11, 1146. [Google Scholar] [CrossRef]
- Yeh, H.-M.; Yu, C.-Y.; Yang, H.-C.; Ko, S.-H.; Liao, C.-L.; Lin, Y.-L. Ubiquitin-Specific Protease 13 Regulates IFN Signaling by Stabilizing STAT1. J. Immunol. 2013, 191, 3328–3336. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Bist, P.; Dikshit, N.; Krishnan, M.N. Global Functional Profiling of Human Ubiquitome Identifies E3 Ubiquitin Ligase DCST1 as a Novel Negative Regulator of Type-I Interferon Signaling. Sci. Rep. 2016, 6, 36179. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-J.; An, H.-J.; Kim, S.-M.; Yoo, S.-M.; Park, J.; Lee, G.-E.; Kim, W.-Y.; Kim, D.J.; Kang, H.C.; Lee, J.Y.; et al. FBXW7-Mediated Stability Regulation of Signal Transducer and Activator of Transcription 2 in Melanoma Formation. Proc. Natl. Acad. Sci. USA 2020, 117, 584–594. [Google Scholar] [CrossRef]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP–Ubiquitin Ligase Complex Associates Specifically with Phosphorylated Destruction Motifs in IκBα and β-Catenin and Stimulates IκBα Ubiquitination in Vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef]
- Spencer, E.; Jiang, J.; Chen, Z.J. Signal-Induced Ubiquitination of IκBα by the F-Box Protein Slimb/β-TrCP. Genes Dev. 1999, 13, 284–294. [Google Scholar] [CrossRef]
- Ji, J.; Ding, K.; Luo, T.; Zhang, X.; Chen, A.; Zhang, D.; Li, G.; Thorsen, F.; Huang, B.; Li, X.; et al. TRIM22 Activates NF-ΚB Signaling in Glioblastoma by Accelerating the Degradation of IκBα. Cell Death Differ. 2021, 28, 367–381. [Google Scholar] [CrossRef]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-Mediated Termination of Transcription Factor NF-KappaB Activation by Intranuclear Sequestration and Degradation of the P65 Subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-KappaB Signaling by Pin1-Dependent Prolyl Isomerization and Ubiquitin-Mediated Proteolysis of P65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Ganesh, L.; Burstein, E.; Guha-Niyogi, A.; Louder, M.K.; Mascola, J.R.; Klomp, L.W.J.; Wijmenga, C.; Duckett, C.S.; Nabel, G.J. The Gene Product Murr1 Restricts HIV-1 Replication in Resting CD4+ Lymphocytes. Nature 2003, 426, 853–857. [Google Scholar] [CrossRef]
- Maine, G.N.; Mao, X.; Komarck, C.M.; Burstein, E. COMMD1 Promotes the Ubiquitination of NF-KappaB Subunits through a Cullin-Containing Ubiquitin Ligase. EMBO J. 2007, 26, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Ito, Y.; Ichikawa, S.; Tokunaga, M.; Sakata-Sogawa, K.; Tanaka, T. MKRN2 Is a Novel Ubiquitin E3 Ligase for the P65 Subunit of NF-ΚB and Negatively Regulates Inflammatory Responses. Sci. Rep. 2017, 7, 46097. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Sun, Y.; Chang, H.; Sun, X.; Yang, S. The E3 Ubiquitin Ligase RNF182 Inhibits TLR-Triggered Cytokine Production through Promoting P65 Ubiquitination and Degradation. FEBS Lett. 2019, 593, 3210–3219. [Google Scholar] [CrossRef]
- Gao, Z.; Li, Y.; Wang, F.; Huang, T.; Fan, K.; Zhang, Y.; Zhong, J.; Cao, Q.; Chao, T.; Jia, J.; et al. Mitochondrial Dynamics Controls Anti-Tumour Innate Immunity by Regulating CHIP-IRF1 Axis Stability. Nat. Commun. 2017, 8, 1805. [Google Scholar] [CrossRef]
- Landré, V.; Pion, E.; Narayan, V.; Xirodimas, D.P.; Ball, K.L. DNA-Binding Regulates Site-Specific Ubiquitination of IRF-1. Biochem. J. 2013, 449, 707–717. [Google Scholar] [CrossRef]
- Higgs, R.; Lazzari, E.; Wynne, C.; Ní Gabhann, J.; Espinosa, A.; Wahren-Herlenius, M.; Jefferies, C.A. Self Protection from Anti-Viral Responses—Ro52 Promotes Degradation of the Transcription Factor IRF7 Downstream of the Viral Toll-Like Receptors. PLoS ONE 2010, 5, e11776. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.-F.; Li, S.; Wang, Z.-X.; Liu, S.-B.; Chen, D.-D.; Zhang, Y.-A. Zebrafish NDRG1a Negatively Regulates IFN Induction by Promoting the Degradation of IRF7. J. Immunol. 2019, 202, 119–130. [Google Scholar] [CrossRef]
- Chase, B.I.; Zhou, Y.; Xiang, Y.; Silverman, R.H.; Zhou, A. Proteasome-Mediated Degradation of RNase L in Response to Phorbol-12-Myristate-13-Acetate (PMA) Treatment of Mouse L929 Cells. J. Interferon Cytokine Res. 2003, 23, 565–573. [Google Scholar] [CrossRef]
- Ma, Q.; Li, J.; Zhou, H.; Tong, W.; Chen, Y. The Function of RNase L and Its Degradation Mechanism in Cardiac Acute Ischemic Injury. Apoptosis 2020, 25, 400–411. [Google Scholar] [CrossRef]
- Yuan, Y.; Miao, Y.; Qian, L.; Zhang, Y.; Liu, C.; Liu, J.; Zuo, Y.; Feng, Q.; Guo, T.; Zhang, L.; et al. Targeting UBE4A Revives Viperin Protein in Epithelium to Enhance Host Antiviral Defense. Mol. Cell 2020, 77, 734–747.e7. [Google Scholar] [CrossRef]
- Chan, Y.-L.; Chang, T.-H.; Liao, C.-L.; Lin, Y.-L. The Cellular Antiviral Protein Viperin Is Attenuated by Proteasome-Mediated Protein Degradation in Japanese Encephalitis Virus-Infected Cells. J. Virol. 2008, 82, 10455–10464. [Google Scholar] [CrossRef]
- Krishnamurthy, P.M.; Shukla, S.; Ray, P.; Mehra, R.; Nyati, M.K.; Lawrence, T.S.; Ray, D. Involvement of P38-ΒTrCP-Tristetraprolin-TNFα Axis in Radiation Pneumonitis. Oncotarget 2017, 8, 47767–47779. [Google Scholar] [CrossRef]
- Ngoc, L.V.; Wauquier, C.; Soin, R.; Bousbata, S.; Twyffels, L.; Kruys, V.; Gueydan, C. Rapid Proteasomal Degradation of Posttranscriptional Regulators of the TIS11/Tristetraprolin Family Is Induced by an Intrinsically Unstructured Region Independently of Ubiquitination. Mol. Cell. Biol. 2014, 34, 4315–4328. [Google Scholar] [CrossRef][Green Version]
- Huang, L.; Yu, Z.; Zhang, Z.; Ma, W.; Song, S.; Huang, G. Interaction with Pyruvate Kinase M2 Destabilizes Tristetraprolin by Proteasome Degradation and Regulates Cell Proliferation in Breast Cancer. Sci. Rep. 2016, 6, 22449. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, C.; Griseri, P.; Grépin, R.; Bertolotto, C.; Mazure, N.; Pagès, G. Constitutive ERK Activity Induces Downregulation of Tristetraprolin, a Major Protein Controlling Interleukin8/CXCL8 MRNA Stability in Melanoma Cells. Am. J. Physiol. Cell Physiol. 2011, 301, C609–C618. [Google Scholar] [CrossRef]
- Brook, M.; Tchen, C.R.; Santalucia, T.; McIlrath, J.; Arthur, J.S.C.; Saklatvala, J.; Clark, A.R. Posttranslational Regulation of Tristetraprolin Subcellular Localization and Protein Stability by P38 Mitogen-Activated Protein Kinase and Extracellular Signal-Regulated Kinase Pathways. Mol. Cell. Biol. 2006, 26, 2408–2418. [Google Scholar] [CrossRef]
- Deleault, K.M.; Skinner, S.J.; Brooks, S.A. Tristetraprolin Regulates TNF TNF-Alpha MRNA Stability via a Proteasome Dependent Mechanism Involving the Combined Action of the ERK and P38 Pathways. Mol. Immunol. 2008, 45, 13–24. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budroni, V.; Versteeg, G.A. Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination. Viruses 2021, 13, 584. https://doi.org/10.3390/v13040584
Budroni V, Versteeg GA. Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination. Viruses. 2021; 13(4):584. https://doi.org/10.3390/v13040584
Chicago/Turabian StyleBudroni, Valentina, and Gijs A. Versteeg. 2021. "Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination" Viruses 13, no. 4: 584. https://doi.org/10.3390/v13040584
APA StyleBudroni, V., & Versteeg, G. A. (2021). Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination. Viruses, 13(4), 584. https://doi.org/10.3390/v13040584