
Contributions of Ubiquitin and Ubiquitination to Flaviviral Antagonism of Type I IFN



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
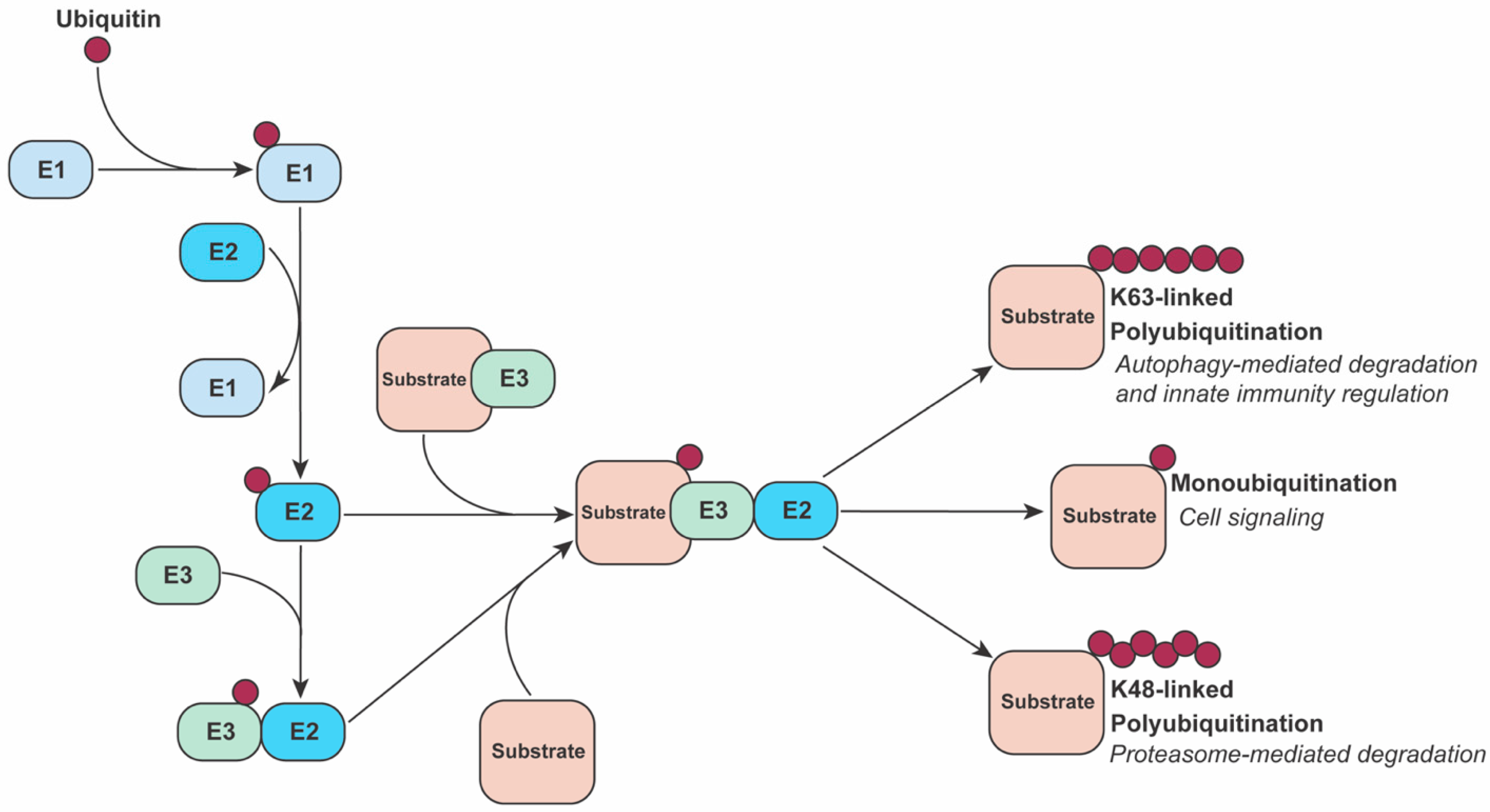
2. The Ubiquitin System
3. The Type I Interferon Response
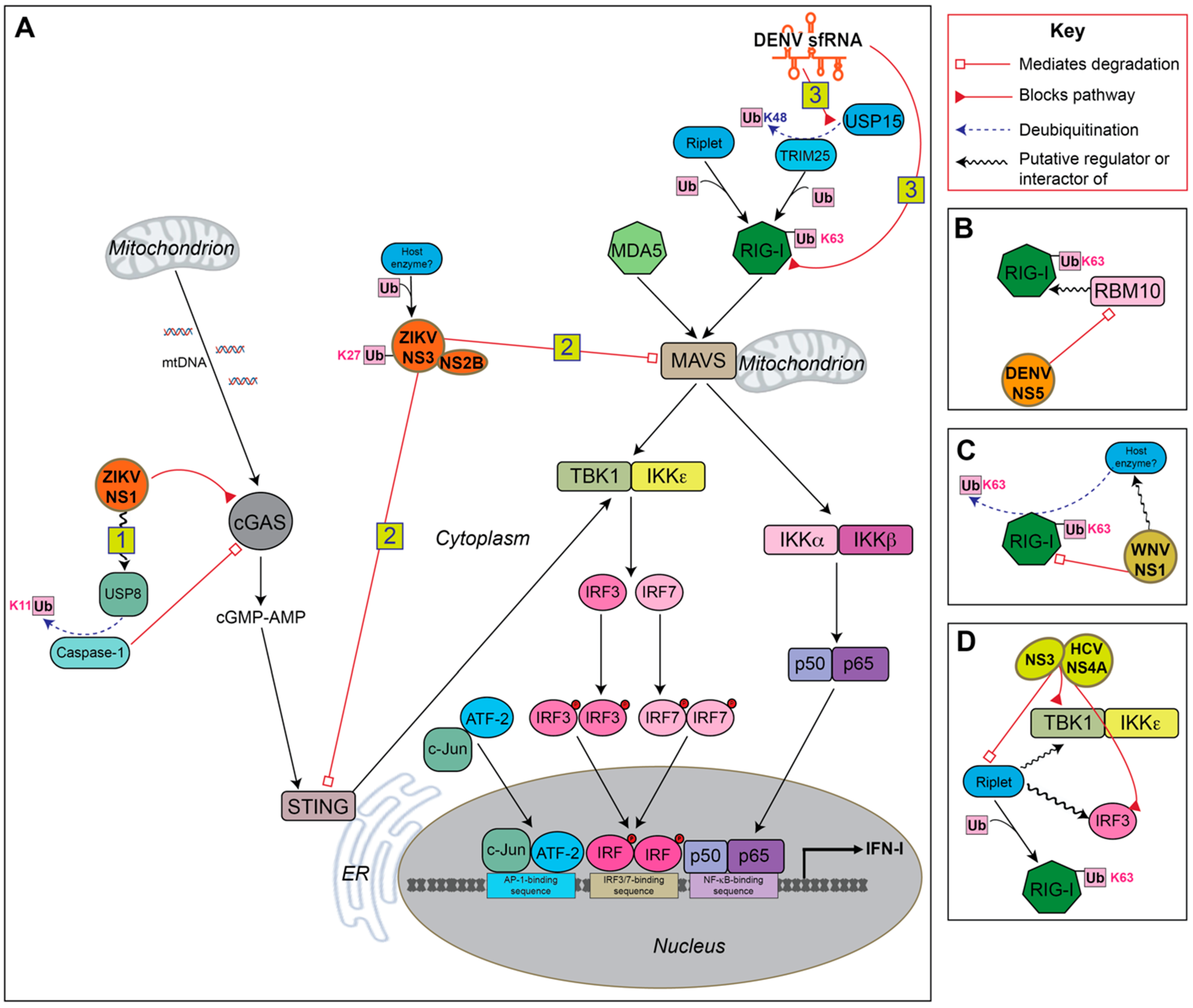
4. Ubiquitin-Mediated Antagonism of IFN-I Production by Flaviviruses
4.1. sfRNA
4.2. NS1
4.3. NS2B, NS3 and NS2B3
4.4. NS5
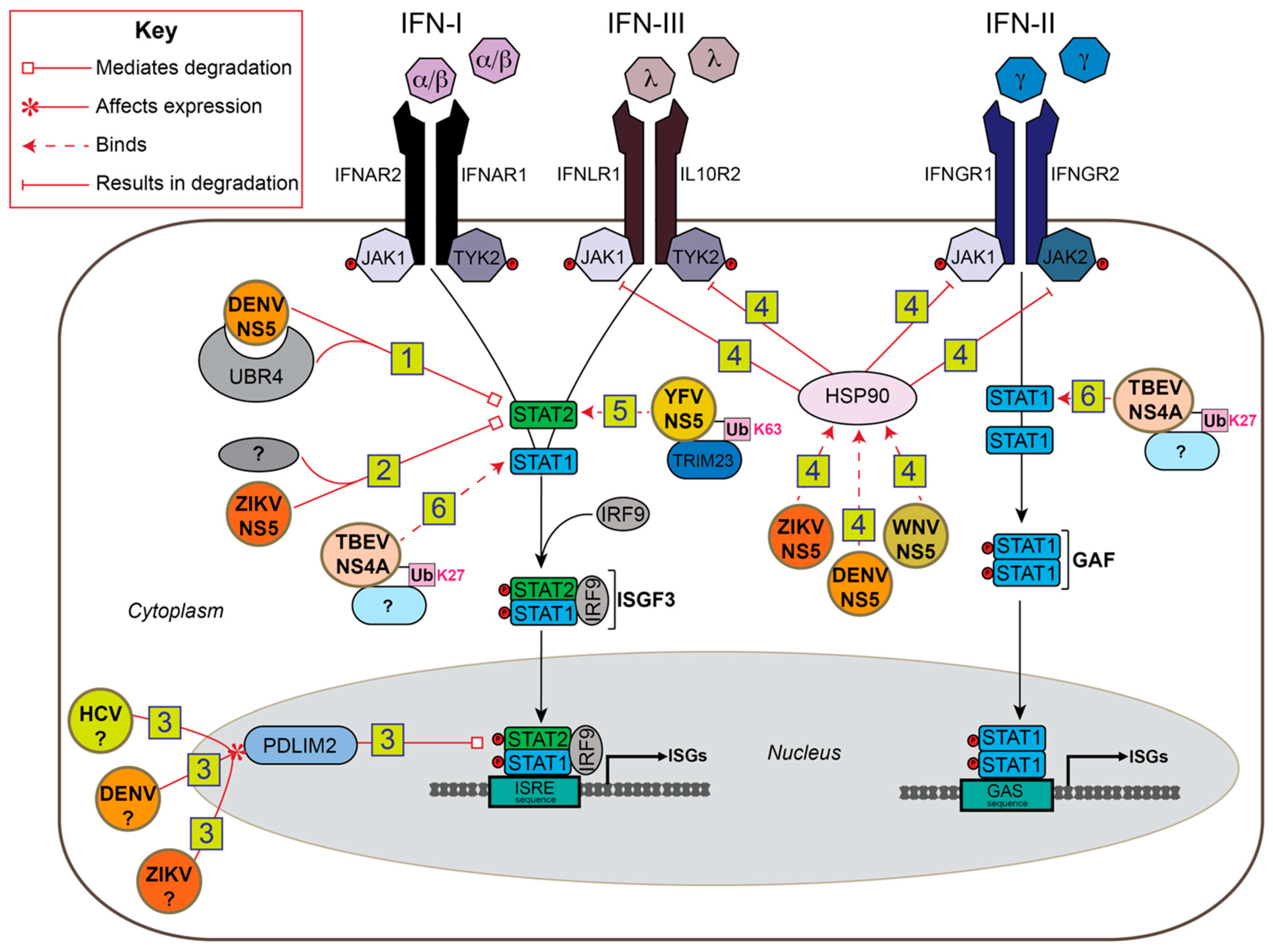
5. Ubiquitin-Mediated Antagonism of IFN-I Signaling by Flaviviruses
5.1. NS5
5.2. NS4A
6. Ubiquitin-Mediated Antagonism of JAK/STAT Signaling in Insects
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gubler, D.J. Dengue/Dengue Haemorrhagic Fever: History and Current Status. Novartis Found. Symp. 2008, 277, 3–22. [Google Scholar] [CrossRef]
- Guarner, J.; Hale, G.L. Four human diseases with significant public health impact caused by mosquito-borne flaviviruses: West Nile, Zika, dengue and yellow fever. Semin. Diagn. Pathol. 2019, 36, 170–176. [Google Scholar] [CrossRef]
- Dub, T.; Ollgren, J.; Huusko, S.; Uusitalo, R.; Siljander, M.; Vapalahti, O.; Sane, J. Game Animal Density, Climate, and Tick-Borne Encephalitis in Finland, 2007–2017. Emerg. Infect. Dis. 2020, 26, 2899–2906. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Guzman, M.G.; Halstead, S.B.; Artsob, H.; Buchy, P.; Farrar, J.; Gubler, D.J.; Hunsperger, E.; Kroeger, A.; Margolis, H.S.; Martínez, E.; et al. Dengue: A continuing global threat. Nat. Rev. Genet. 2010, 8, S7–S16. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Lira-Vieira, A.R.; Gurgel-Gonçalves, R.; Moreira, I.M.; Yoshizawa, M.A.C.; Coutinho, M.L.; Prado, P.S.; De Souza, J.L.; Chaib, A.J.D.M.; Moreira, J.S.; De Castro, C.N. Ecological aspects of mosquitoes (Diptera: Culicidae) in the gallery forest of Brasilia National Park, Brazil, with an emphasis on potential vectors of yellow fever. Rev. Soc. Bras. Med. Trop. 2013, 46, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Liao, K.-C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.-C.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef] [PubMed]
- Lubick, K.J.; Robertson, S.J.; McNally, K.L.; Freedman, B.A.; Rasmussen, A.L.; Taylor, R.T.; Walts, A.D.; Tsuruda, S.; Sakai, M.; Ishizuka, M.; et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe 2015, 18, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain Is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.-Y.; Garcia-Sastre, A. NS5 of Dengue Virus Mediates STAT2 Binding and Degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue Virus NS5 Inhibits Interferon-α Signaling by Blocking Signal Transducer and Activator of Transcription 2 Phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.F.; Fernandez-Sesma, A.; García-Sastre, A. Dengue Virus Co-opts UBR4 to Degrade STAT2 and Antagonize Type I Interferon Signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Morrison, J.; Laurent-Rolle, M.; Belicha-Villanueva, A.; Plumlee, C.R.; Bernal-Rubio, D.; Williams, K.L.; Harris, E.; Fernandez-Sesma, A.; Schindler, C.; et al. Mouse STAT2 Restricts Early Dengue Virus Replication. Cell Host Microbe 2010, 8, 410–421. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; MacLeod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martínez-Romero, C.; Tenoever, B.R.; et al. The Interferon Signaling Antagonist Function of Yellow Fever Virus NS5 Protein Is Activated by Type I Interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef]
- Lin, R.-J.; Chang, B.-L.; Yu, H.-P.; Liao, C.-L.; Lin, Y.-L. Blocking of Interferon-Induced Jak-Stat Signaling by Japanese Encephalitis Virus NS5 through a Protein Tyrosine Phosphatase-Mediated Mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef]
- Yang, T.-C.; Li, S.-W.; Lai, C.-C.; Lu, K.-Z.; Chiu, M.-T.; Hsieh, T.-H.; Wan, L.; Lin, C.-W. Proteomic analysis for Type I interferon antagonism of Japanese encephalitis virus NS5 protein. Proteomics 2013, 13, 3442–3456. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of Interferon-Stimulated JAK-STAT Signaling by a Tick-Borne Flavivirus and Identification of NS5 as an Interferon Antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef] [PubMed]
- Park, G.S.; Morris, K.L.; Hallett, R.G.; Bloom, M.E.; Best, S.M. Identification of Residues Critical for the Interferon Antagonist Function of Langat Virus NS5 Reveals a Role for the RNA-Dependent RNA Polymerase Domain. J. Virol. 2007, 81, 6936–6946. [Google Scholar] [CrossRef]
- Valmas, C.; Grosch, M.N.; Schümann, M.; Olejnik, J.; Martinez, O.; Best, S.M.; Krähling, V.; Basler, C.F.; Mühlberger, E. Marburg Virus Evades Interferon Responses by a Mechanism Distinct from Ebola Virus. PLoS Pathog. 2010, 6, e1000721. [Google Scholar] [CrossRef]
- Werme, K.; Wigerius, M.; Johansson, M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 2008, 10, 696–712. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Duchemin, J.-B.; Rodriguez-Andres, J.; Trinidad, L.; Walker, P.J. Cullin4 Is Pro-Viral during West Nile Virus Infection of Culex Mosquitoes. PLoS Pathog. 2015, 11, e1005143. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Laurent-Rolle, M.; Pisanelli, G.; Co, P.H.; Albrecht, R.A.; García-Sastre, A.; Morrison, J. Host-Specific NS5 Ubiquitination Determines Yellow Fever Virus Tropism. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Yuen, K.-S.; Chan, J.F.-W.; Chan, C.-P.; Wang, P.-H.; Cai, J.-P.; Zhang, S.; Liang, M.; Kok, K.-H.; Yuen, K.-Y.; et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J. Virol. 2017, 91, e00163-17. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chen, Z.; Li, Y.; Zhao, Z.; He, W.; Zohaib, A.; Cao, S. Japanese Encephalitis Virus NS5 Inhibits Type I Interferon (IFN) Production by Blocking the Nuclear Translocation of IFN Regulatory Factor 3 and NF-kappaB. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Ciechanover, A.; Varshavsky, A. Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell 1984, 37, 43–55. [Google Scholar] [CrossRef]
- Hershko, A.; Leshinsky, E.; Ganoth, D.; Heller, H. ATP-dependent degradation of ubiquitin-protein conjugates. Proc. Natl. Acad. Sci. USA 1984, 81, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The early history of the ubiquitin field. Protein Sci. 2006, 15, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Cagno, V.; Tseligka, E.D.; Bettex, Q.; Huang, S.; Constant, S.; Tapparel, C. Growth of Zika virus in human reconstituted respiratory, intestinal, vaginal and neural tissues. Clin. Microbiol. Infect. 2019, 25, 1042.e1–1042.e4. [Google Scholar] [CrossRef] [PubMed]
- Gustin, J.K.P.; Moses, A.V.P.; Früh, K.P.; Douglas, J.L.P. Viral Takeover of the Host Ubiquitin System. Front. Microbiol. 2011, 2, 161. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Henrici, R.C.; Tan, S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nat. Cell Biol. 2014, 514, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin enzymes in the regulation of immune responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 425–460. [Google Scholar] [CrossRef] [PubMed]
- Locke, M.; Toth, J.I.; Petroski, M.D. Lys11- and Lys48-linked ubiquitin chains interact with p97 during endoplasmic-reticulum-associated degradation. Biochem. J. 2014, 459, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623. [Google Scholar] [CrossRef]
- Matsumoto, M.L.; Wickliffe, K.E.; Dong, K.C.; Yu, C.; Bosanac, I.; Bustos, D.; Phu, L.; Kirkpatrick, D.S.; Hymowitz, S.G.; Rape, M.; et al. K11-Linked Polyubiquitination in Cell Cycle Control Revealed by a K11 Linkage-Specific Antibody. Mol. Cell 2010, 39, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Van Huizen, M.; Kikkert, M. The Role of Atypical Ubiquitin Chains in the Regulation of the Antiviral Innate Immune Response. Front. Cell Dev. Biol. 2019, 7, 392. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhou, M.-T.; Hu, M.-M.; Hu, Y.-H.; Zhang, J.; Guo, L.; Zhong, B.; Shu, H.-B. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PLoS Pathog. 2014, 10, e1004358. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R. USP 19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 2016, 35, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Kensche, T.; Tokunaga, F.; Ikeda, F.; Goto, E.; Iwai, K.; Dikic, I. Analysis of nuclear factor-kappaB (NF-kappaB) essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-kappaB. J. Biol. Chem. 2012, 287, 23626–23634. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Paz, S.; Samuel, S.; Goulet, M.-L.; Sun, Q.; Kikkert, M.; Iwai, K.; Dikic, I.; Hiscott, J.; Lin, R. Linear Ubiquitination of NEMO Negatively Regulates the Interferon Antiviral Response through Disruption of the MAVS-TRAF3 Complex. Cell Host Microbe 2012, 12, 211–222. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, M.; Wang, W.; Liu, W.; Song, X.; Ma, Z.; Zhang, S.; Liu, L.; Liu, Y.; Cao, X. Nuclear RNF2 inhibits interferon function by promoting K33-linked STAT1 disassociation from DNA. Nat. Immunol. 2018, 19, 41–52. [Google Scholar] [CrossRef]
- Lin, M.; Zhao, Z.; Yang, Z.; Meng, Q.; Tan, P.; Xie, W.; Qin, Y.; Wang, R.-F.; Cui, J. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination through NLRP4 Signalosome. Mol. Cell 2016, 64, 267–281. [Google Scholar] [CrossRef]
- Lei, C.Q.; Wu, X.; Zhong, X.; Jiang, L.; Zhong, B.; Shu, H.B. USP19 Inhibits TNF-alpha- and IL-1beta-Triggered NF-kappaB Activation by Deubiquitinating TAK1. J. Immunol. 2019, 203, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Jin, J.; Luan, Y.; Chen, C.; Li, Y.; Chu, H.; Wang, X.; Liao, G.; Yu, Y.; et al. p38 inhibition provides anti–DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J. Exp. Med. 2017, 214, 991–1010. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Q.; Jing, Y.-Y.; Zhang, M.; Wang, H.-Y.; Cai, Z.; Liuyu, T.; Zhang, Z.-D.; Xiong, T.-C.; Wu, Y.; et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat. Commun. 2017, 8, 15534. [Google Scholar] [CrossRef]
- Liu, J.; Han, C.; Xie, B.; Wu, Y.; Liu, S.; Chen, K.; Xia, M.; Zhuang, Y.; Song, L.; Li, Z.; et al. Rhbdd3 controls autoimmunity by suppressing the production of IL-6 by dendritic cells via K27-linked ubiquitination of the regulator NEMO. Nat. Immunol. 2014, 15, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.-I.; Funami, K.; Saeki, Y.; Tanaka, K.; Okawa, K.; Takeuchi, O.; Akira, S.; Murakami, Y.; Shimotohno, K. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. USA 2010, 107, 15856–15861. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, L.; Hong, Z.; Lv, Z.; Mao, Z.; Tang, Y.; Zhou, Q. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017, 13, e1006264. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92, JVI.00321-18. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Hertzog, P.J.; Holland, K.A.; Sumarsono, S.H.; Tymms, M.J.; Hamilton, J.A.; Whitty, G.; Bertoncello, I.; Kola, I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc. Natl. Acad. Sci. USA 1995, 92, 11284–11288. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.; Steinhoff, U.; Reis, L.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Hendriks, W.; Althage, A.; Hemmi, S.; Bluethmann, H.; Kamijo, R.; Vilcek, J.; Zinkernagel, R.; Aguet, M. Immune response in mice that lack the interferon-gamma receptor. Science 1993, 259, 1742–1745. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Boil. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Dalton, D.; Pitts-Meek, S.; Keshav, S.; Figari, I.; Bradley, A.; Stewart, T. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 1993, 259, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Severa, M.; Giacomini, E.; Monneron, D.; Remoli, M.E.; Julkunen, I.; Uzé, G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. Overview of the biology of type I interferons. Arthritis Res. Ther. 2010, 12, S1. [Google Scholar] [CrossRef]
- Bekisz, J.; Schmeisser, H.; Hernandez, J.; Goldman, N.D.; Zoon, K.C. Mini ReviewHuman Interferons Alpha, Beta and Omega. Growth Factors 2004, 22, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; E Sousa, C.R. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5’-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nat. Cell Biol. 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Sun, X.; Xian, H.; Tian, S.; Sun, T.; Qin, Y.; Zhang, S.; Cui, J. A Hierarchical Mechanism of RIG-I Ubiquitination Provides Sensitivity, Robustness and Synergy in Antiviral Immune Responses. Sci. Rep. 2016, 6, 29263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Olagnier, D.; Lin, R. Host and Viral Modulation of RIG-I-Mediated Antiviral Immunity. Front. Immunol. 2017, 7, 662. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nat. Cell Biol. 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Kirchhofer, A.; Shin, Y.C.; Inn, K.-S.; Liang, C.; Cui, S.; Myong, S.; Ha, T.; Hopfner, K.-P.; Jung, J.U. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc. Natl. Acad. Sci. USA 2008, 105, 16743–16748. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Xie, W.; Yang, S.; Liu, Q.; Xia, X.; Jin, S.; Sun, T.; Cui, J. Stratified ubiquitination of RIG-I creates robust immune response and induces selective gene expression. Sci. Adv. 2017, 3, e1701764. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nat. Cell Biol. 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS 1-caspase-1 axis. EMBO J. 2018, 37, e99347. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, N.; Dai, S.; Hou, G.; Guo, K.; Chen, X.; Yi, C.; Liu, W.; Deng, F.; Wu, Y.; et al. Zika virus circumvents host innate immunity by targeting the adaptor proteins MAVS and MITA. FASEB J. 2019, 33, 9929–9944. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, B.; Bragado, L.; Mammi, P.; Torti, M.F.; Gaioli, N.; Gebhard, L.G.; Solá, M.E.G.; Vaz-Drago, R.; Iglesias, N.G.; García, C.C.; et al. Dengue virus targets RBM10 deregulating host cell splicing and innate immune response. Nucleic Acids Res. 2020, 48, 6824–6838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-L.; Ye, H.-Q.; Liu, S.-Q.; Deng, C.-L.; Li, X.-D.; Shi, P.-Y.; Zhang, B. West Nile Virus NS1 Antagonizes Interferon Beta Production by Targeting RIG-I and MDA5. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A Distinct Role of Riplet-Mediated K63-Linked Polyubiquitination of the RIG-I Repressor Domain in Human Antiviral Innate Immune Responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Tan, C.Y.; Horner, S.M. Hepatitis C Virus Infection Is Inhibited by a Noncanonical Antiviral Signaling Pathway Targeted by NS3-NS4A. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Yang, Y.K.; Wang, R.P.; Zhou, X.; Diao, F.C.; Li, M.D.; Chen, D.Y. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 2009, 4, e5760. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The Ubiquitin Ligase Riplet Is Essential for RIG-I-Dependent Innate Immune Responses to RNA Virus Infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; I Webb, A.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.-W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187–1200.e16. [Google Scholar] [CrossRef]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef]
- Hiscott, J. Triggering the Innate Antiviral Response through IRF-3 Activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658. [Google Scholar] [CrossRef]
- Lai, J.; Wang, M.; Huang, C.; Wu, C.; Hung, L.; Yang, C.; Ke, P.; Luo, S.; Liu, S.; Ho, L. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018, 19, e46182. [Google Scholar] [CrossRef]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Berry-Wynne, K.M.; Dos Santos, T.; Addison, W.R.; McFarlane, N.; Hobman, T.; Tyrrell, D.L. HCV and flaviviruses hijack cellular mechanisms for nuclear STAT2 degradation: Up-regulation of PDLIM2 suppresses the innate immune response. PLoS Pathog. 2019, 15, e1007949. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Esser-Nobis, K.; Dewey-Verstelle, E.C.; Fairgrieve, M.R.; Schwerk, J.; Lu, A.Y.; Soveg, F.W.; Hemann, E.A.; Hatfield, L.D.; Keller, B.C.; et al. Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling. Cells 2020, 9, 899. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; You, J.; Zhou, Y.; Wang, Y.; Pei, R.; Chen, X.; Chen, J. Tick-borne encephalitis virus NS4A ubiquitination antagonizes type I interferon-stimulated STAT1/2 signalling pathway. Emerg. Microbes. Infect. 2020, 9, 714–726. [Google Scholar] [CrossRef]
- Becquart, P.; Wauquier, N.; Nkoghe, D.; Ndjoyi-Mbiguino, A.; Padilla, C.; Souris, M.; Leroy, E.M. Acute dengue virus 2 infection in Gabonese patients is associated with an early innate immune response, including strong interferon alpha production. BMC Infect. Dis. 2010, 10, 356. [Google Scholar] [CrossRef]
- Lobigs, M.; Müllbacher, A.; Wang, Y.; Pavy, M.; Lee, E. Role of type I and type II interferon responses in recovery from infection with an encephalitic flavivirus. J. Gen. Virol. 2003, 84, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/Beta Interferon Protects against Lethal West Nile Virus Infection by Restricting Cellular Tropism and Enhancing Neuronal Survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed]
- Shresta, S.; Kyle, J.L.; Snider, H.M.; Basavapatna, M.; Beatty, P.R.; Harris, E. Interferon-Dependent Immunity Is Essential for Resistance to Primary Dengue Virus Infection in Mice, Whereas T- and B-Cell-Dependent Immunity Are Less Critical. J. Virol. 2004, 78, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xing, Y.; Chen, X.; Zheng, Y.; Yang, Y.; Nichols, D.B.; Clementz, M.A.; Banach, B.S.; Li, K.; Baker, S.C.; et al. Coronavirus Papain-like Proteases Negatively Regulate Antiviral Innate Immune Response through Disruption of STING-Mediated Signaling. PLoS ONE 2012, 7, e30802. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.-K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The Ubiquitin-Specific Protease USP15 Promotes RIG-I-Mediated Antiviral Signaling by Deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, K.; Huang, Y.; Sun, M.; Tian, Q.; Zhang, S.; Qin, Y. TRIM25 Promotes TNF-alpha-Induced NF-kappaB Activation through Potentiating the K63-Linked Ubiquitination of TRAF2. J. Immunol. 2020, 204, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef]
- Schuessler, A.; Funk, A.; LaZear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile Virus Noncoding Subgenomic RNA Contributes to Viral Evasion of the Type I Interferon-Mediated Antiviral Response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef] [PubMed]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; França, R.F.D.O.; Pena, L.J.; Wilkie, G.S.; Filipe, A.D.S.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Neglected Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nat. Cell Biol. 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, L.; Sun, J.; Chen, W.; Li, S.; Wang, Q.; Yu, H.; Xia, Z.; Jin, X.; Wang, C. Endoplasmic Reticulum Protein SCAP Inhibits Dengue Virus NS2B3 Protease by Suppressing Its K27-Linked Polyubiquitylation. J. Virol. 2017, 91, e02234-16. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; Ploss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef]
- Stabell, A.C.; Meyerson, N.R.; Gullberg, R.C.; Gilchrist, A.R.; Webb, K.J.; Old, W.M.; Perera, R.; Sawyer, S.L. Dengue viruses cleave STING in humans but not in nonhuman primates, their presumed natural reservoir. eLife 2018, 7, e31919. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-linked Ubiquitination*. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Tu, D.; Zhu, Z.; Zhou, A.Y.; Yun, C.-H.; Lee, K.-E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and Ubiquitination-Dependent Activation of TANK-Binding Kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Keinanen, T.A.; Ivanova, O.N.; Hyvonen, M.T.; Khomutov, A.R.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Hepatitis C virus alters metabolism of biogenic polyamines by affecting expression of key enzymes of their metabolism. Biochem. Biophys. Res. Commun. 2017, 483, 904–909. [Google Scholar] [CrossRef]
- Tasaki, T.; Mulder, L.C.F.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A Family of Mammalian E3 Ubiquitin Ligases That Contain the UBR Box Motif and Recognize N-Degrons. Mol. Cell. Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Soriano, M.A.; Grusby, M.J. SLIM Is a Nuclear Ubiquitin E3 Ligase that Negatively Regulates STAT Signaling. Immunity 2005, 22, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Mi, Z.; Bowles, D.E.; Bhattacharya, S.D.; Kuo, P.C. Osteopontin and Protein Kinase C Regulate PDLIM2 Activation and STAT1 Ubiquitination in LPS-treated Murine Macrophages. J. Biol. Chem. 2010, 285, 37787–37796. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Healy, N.C.; O’Connor, R. Sequestration of PDLIM2 in the cytoplasm of monocytic/macrophage cells is associated with adhesion and increased nuclear activity of NF-kappaB. J. Leukoc. Biol. 2009, 85, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Fragkoudis, R.; Attarzadeh-Yazdi, G.; Nash, A.A.; Fazakerley, J.K.; Kohl, A. Advances in dissecting mosquito innate immune responses to arbovirus infection. J. Gen. Virol. 2009, 90, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Trinidad, L.; Voysey, R.; Duchemin, J.-B.; Walker, P.J. Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 18915–18920. [Google Scholar] [CrossRef]
- Souza-Neto, J.A.; Sim, S.; Dimopoulos, G. An evolutionary conserved function of the JAK-STAT pathway in anti-dengue defense. Proc. Natl. Acad. Sci. USA 2009, 106, 17841–17846. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hay-McCullough, E.; Morrison, J. Contributions of Ubiquitin and Ubiquitination to Flaviviral Antagonism of Type I IFN. Viruses 2021, 13, 763. https://doi.org/10.3390/v13050763
Hay-McCullough E, Morrison J. Contributions of Ubiquitin and Ubiquitination to Flaviviral Antagonism of Type I IFN. Viruses. 2021; 13(5):763. https://doi.org/10.3390/v13050763
Chicago/Turabian StyleHay-McCullough, Erika, and Juliet Morrison. 2021. "Contributions of Ubiquitin and Ubiquitination to Flaviviral Antagonism of Type I IFN" Viruses 13, no. 5: 763. https://doi.org/10.3390/v13050763
APA StyleHay-McCullough, E., & Morrison, J. (2021). Contributions of Ubiquitin and Ubiquitination to Flaviviral Antagonism of Type I IFN. Viruses, 13(5), 763. https://doi.org/10.3390/v13050763