
Novel Genetic Rearrangements in Hepatitis B Virus: Complex Structural Variations and Structural Variation Polymorphisms

Abstract
1. Introduction
2. Complex Structural Variation in the Cellular Genomes of Humans and Mice
3. Complex SVs Observed in the HBV Genome
4. Bioinformatical Analysis for the Detection of Complex SVs in HBV
5. Points Where Complex SVs Are Observed in the HBV Genome
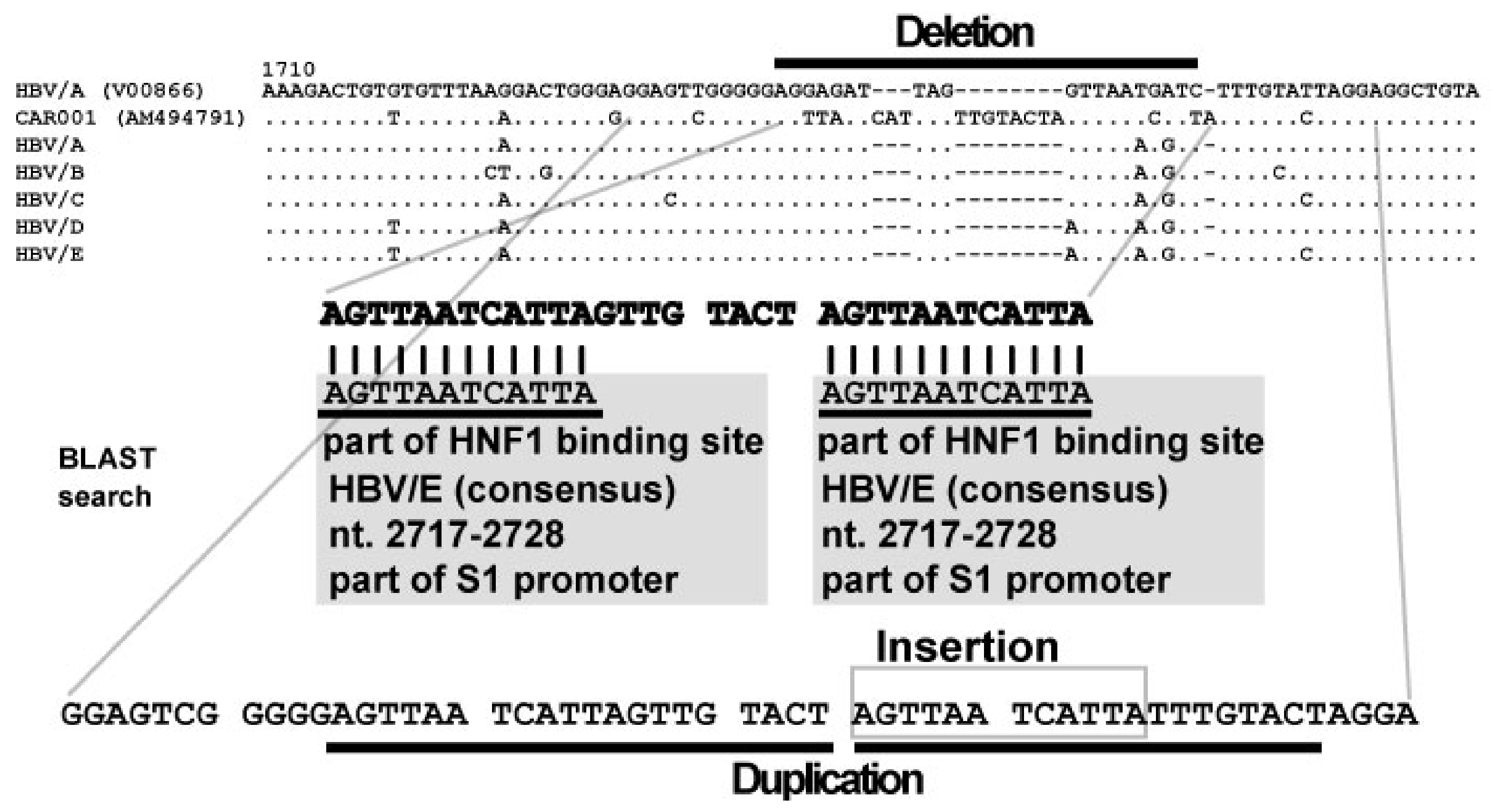
6. Discovery of Insertional Motif Sequences in Complex SVs in HBV
7. Classification of Complex SVs in HBV
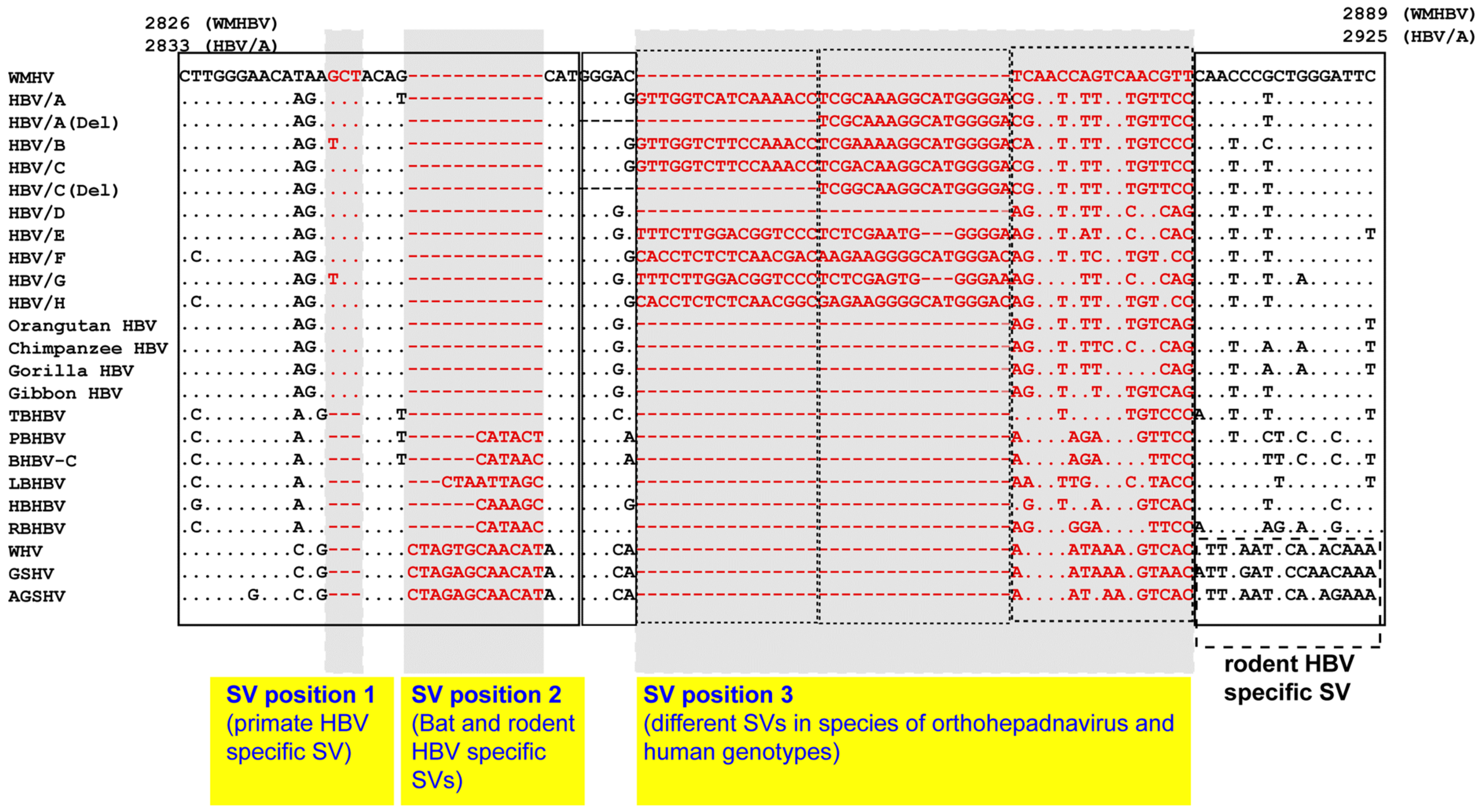
8. SV Polymorphisms
8.1. SV Polymorphisms
8.2. SV Polymorphisms in the Core Region
8.3. SV Polymorphisms in the Pre-S1 Region
8.4. Clinical and Scientific Perspectives on Complex SVs and SV Polymorphisms
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability
Conflicts of Interest
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015. [Google Scholar] [CrossRef]
- Blumberg, B.S.; Alter, H.J.; Visnich, S. A “New” Antigen in Leukemia Sera. JAMA 1965, 191, 541–546. [Google Scholar] [CrossRef]
- Galibert, F.; Mandart, E.; Fitoussi, F.; Tiollais, P.; Charnay, P. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 1979, 281, 646–650. [Google Scholar] [CrossRef]
- Pasek, M.; Goto, T.; Gilbert, W.; Zink, B.; Schaller, H.; MacKay, P.; Leadbetter, G.; Murray, K. Hepatitis B virus genes and their expression in E. coli. Nature 1979, 282, 575–579. [Google Scholar] [CrossRef]
- Valenzuela, P.; Gray, P.; Quiroga, M.; Zaldivar, J.; Goodman, H.M.; Rutter, W.J. Nucleotide sequence of the gene coding for the major protein of hepatitis B virus surface antigen. Nature 1979, 280, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Tsuda, F.; Sakugawa, H.; Sastrosoewignjo, R.I.; Imai, M.; Miyakawa, Y.; Mayumi, M. Typing hepatitis B virus by homology in nucleotide sequence: Comparison of surface antigen subtypes. J. Gen. Virol. 1988, 69 Pt 10, 2575–2583. [Google Scholar] [CrossRef]
- Norder, H.; Hammas, B.; Lofdahl, S.; Courouce, A.M.; Magnius, L.O. Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of the corresponding hepatitis B virus strains. J. Gen. Virol. 1992, 73 Pt 5, 1201–1208. [Google Scholar] [CrossRef]
- Arauz-Ruiz, P.; Norder, H.; Robertson, B.H.; Magnius, L.O. Genotype H: A new Amerindian genotype of hepatitis B virus revealed in Central America. J. Gen. Virol. 2002, 83, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Maisonneuve, P.; Bruno, S.; Mondelli, M.U. Is response to antiviral treatment influenced by hepatitis B virus genotype? J. Hepatol. 2010, 52, 441–449. [Google Scholar] [CrossRef]
- Tran, T.T.; Trinh, T.N.; Abe, K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J. Virol. 2008, 82, 5657–5663. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef]
- Carman, W.F.; Jacyna, M.R.; Hadziyannis, S.; Karayiannis, P.; McGarvey, M.J.; Makris, A.; Thomas, H.C. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 1989, 2, 588–591. [Google Scholar] [CrossRef]
- Okamoto, H.; Tsuda, F.; Akahane, Y.; Sugai, Y.; Yoshiba, M.; Moriyama, K.; Tanaka, T.; Miyakawa, Y.; Mayumi, M. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J. Virol. 1994, 68, 8102–8110. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Rambaut, A.; Harvey, P.H.; Holmes, E.C. Recombination between sequences of hepatitis B virus from different genotypes. J. Mol. Evol. 1996, 42, 97–102. [Google Scholar] [CrossRef]
- Bowyer, S.M.; Sim, J.G. Relationships within and between genotypes of hepatitis B virus at points across the genome: Footprints of recombination in certain isolates. J. Gen. Virol. 2000, 81, 379–392. [Google Scholar] [CrossRef]
- Morozov, V.; Pisareva, M.; Groudinin, M. Homologous recombination between different genotypes of hepatitis B virus. Gene 2000, 260, 55–65. [Google Scholar] [CrossRef]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef]
- Suwannakarn, K.; Tangkijvanich, P.; Theamboonlers, A.; Abe, K.; Poovorawan, Y. A novel recombinant of Hepatitis B virus genotypes G and C isolated from a Thai patient with hepatocellular carcinoma. J. Gen. Virol. 2005, 86, 3027–3030. [Google Scholar] [CrossRef] [PubMed]
- Araujo, N.M. Hepatitis B virus intergenotypic recombinants worldwide: An overview. Infect. Genet. Evol. 2015, 36, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Sugauchi, F.; Orito, E.; Ichida, T.; Kato, H.; Sakugawa, H.; Kakumu, S.; Ishida, T.; Chutaputti, A.; Lai, C.L.; Ueda, R.; et al. Hepatitis B virus of genotype B with or without recombination with genotype C over the precore region plus the core gene. J. Virol. 2002, 76, 5985–5992. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef]
- Zhang, F.; Khajavi, M.; Connolly, A.M.; Towne, C.F.; Batish, S.D.; Lupski, J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009, 41, 849–853. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Clark, R.A.; Sokolova, S.; Leibowitz, M.L.; Zhang, Y.; Hurles, M.E.; Mell, J.C.; Hall, I.M. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010, 20, 623–635. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 2012, 28, 43–53. [Google Scholar] [CrossRef]
- Yalcin, B.; Wong, K.; Bhomra, A.; Goodson, M.; Keane, T.M.; Adams, D.J.; Flint, J. The fine-scale architecture of structural variants in 17 mouse genomes. Genome Biol. 2012, 13, R18. [Google Scholar] [CrossRef]
- Guo, Y.; Gu, X.; Sheng, Z.; Wang, Y.; Luo, C.; Liu, R.; Qu, H.; Shu, D.; Wen, J.; Crooijmans, R.P.; et al. A Complex Structural Variation on Chromosome 27 Leads to the Ectopic Expression of HOXB8 and the Muffs and Beard Phenotype in Chickens. PLoS Genet. 2016, 12, e1006071. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef]
- Spies, N.; Weng, Z.; Bishara, A.; McDaniel, J.; Catoe, D.; Zook, J.M.; Salit, M.; West, R.B.; Batzoglou, S.; Sidow, A. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 2017, 14, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Tanaka, Y.; Paulon, E.; Orito, E.; Sugiyama, M.; Ito, K.; Ueda, R.; Mizokami, M.; Naoumov, N.V. Novel type of hepatitis B virus mutation: Replacement mutation involving a hepatocyte nuclear factor 1 binding site tandem repeat in chronic hepatitis B virus genotype E. J. Virol. 2005, 79, 14404–14410. [Google Scholar] [CrossRef][Green Version]
- Fujiwara, K.; Matsunami, K.; Iio, E.; Nojiri, S.; Joh, T. Novel non-canonical genetic rearrangements termed “complex structural variations” in HBV genome. Virus Res. 2017, 238, 84–93. [Google Scholar] [CrossRef]
- Fujiwara, K.; Matsuura, K.; Matsunami, K.; Iio, E.; Nojiri, S. Characterization of hepatitis B virus with complex structural variations. BMC Microbiol. 2018, 18, 202. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Hino, O.; Tabata, S.; Hotta, Y. Evidence for increased in vitro recombination with insertion of human hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1991, 88, 9248–9252. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Peneau, C.; Imbeaud, S.; La Bella, T.; Hirsch, T.Z.; Caruso, S.; Calderaro, J.; Paradis, V.; Blanc, J.F.; Letouze, E.; Nault, J.C.; et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 2021. [Google Scholar] [CrossRef]
- Li, M.; Du, M.; Cong, H.; Gu, Y.; Fang, Y.; Li, J.; Gan, Y.; Tu, H.; Gu, J.; Xia, Q. Characterization of hepatitis B virus DNA integration patterns in intrahepatic cholangiocarcinoma. Hepatol. Res. 2021, 51, 102–115. [Google Scholar] [CrossRef]
- Courtois, G.; Morgan, J.G.; Campbell, L.A.; Fourel, G.; Crabtree, G.R. Interaction of a liver-specific nuclear factor with the fibrinogen and alpha 1-antitrypsin promoters. Science 1987, 238, 688–692. [Google Scholar] [CrossRef]
- Courtois, G.; Baumhueter, S.; Crabtree, G.R. Purified hepatocyte nuclear factor 1 interacts with a family of hepatocyte-specific promoters. Proc. Natl. Acad. Sci. USA 1988, 85, 7937–7941. [Google Scholar] [CrossRef]
- Cereghini, S.; Yaniv, M.; Cortese, R. Hepatocyte dedifferentiation and extinction is accompanied by a block in the synthesis of mRNA coding for the transcription factor HNF1/LFB1. EMBO J. 1990, 9, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.F. Transcriptional activators in hepatocytes. Cell Growth Differ. 1990, 1, 47–52. [Google Scholar] [PubMed]
- Locker, J.; Ghosh, D.; Luc, P.V.; Zheng, J. Definition and prediction of the full range of transcription factor binding sites--The hepatocyte nuclear factor 1 dimeric site. Nucleic Acids Res. 2002, 30, 3809–3817. [Google Scholar] [CrossRef]
- Chang, H.K.; Wang, B.Y.; Yuh, C.H.; Wei, C.L.; Ting, L.P. A liver-specific nuclear factor interacts with the promoter region of the large surface protein gene of human hepatitis B virus. Mol. Cell Biol. 1989, 9, 5189–5197. [Google Scholar] [CrossRef]
- Raney, A.K.; Milich, D.R.; Easton, A.J.; McLachlan, A. Differentiation-specific transcriptional regulation of the hepatitis B virus large surface antigen gene in human hepatoma cell lines. J. Virol. 1990, 64, 2360–2368. [Google Scholar] [CrossRef]
- Zhou, D.X.; Yen, T.S. The ubiquitous transcription factor Oct-1 and the liver-specific factor HNF-1 are both required to activate transcription of a hepatitis B virus promoter. Mol. Cell Biol. 1991, 11, 1353–1359. [Google Scholar] [CrossRef]
- Nishizono, A.; Maeno, M.; Hiraga, M.; Hirai, H.; Esumi, M.; Shikata, T. In vitro transcription of the hepatitis B virus gene by nuclear extracts of human hepatoma cells. Virology 1991, 182, 545–552. [Google Scholar] [CrossRef]
- Raney, A.K.; Easton, A.J.; McLachlan, A. Characterization of the minimal elements of the hepatitis B virus large surface antigen promoter. J. Gen. Virol. 1994, 75 Pt 10, 2671–2679. [Google Scholar] [CrossRef]
- Gunther, S.; Piwon, N.; Iwanska, A.; Schilling, R.; Meisel, H.; Will, H. Type, prevalence, and significance of core promoter/enhancer II mutations in hepatitis B viruses from immunosuppressed patients with severe liver disease. J. Virol. 1996, 70, 8318–8331. [Google Scholar] [CrossRef]
- Pult, I.; Chouard, T.; Wieland, S.; Klemenz, R.; Yaniv, M.; Blum, H.E. A hepatitis B virus mutant with a new hepatocyte nuclear factor 1 binding site emerging in transplant-transmitted fulminant hepatitis B. Hepatology 1997, 25, 1507–1515. [Google Scholar] [CrossRef]
- Laskus, T.; Rakela, J.; Steers, J.L.; Wiesner, R.H.; Persing, D.H. Precore and contiguous regions of hepatitis B virus in liver transplantation for end-stage hepatitis B. Gastroenterology 1994, 107, 1774–1780. [Google Scholar] [CrossRef]
- Gerolami, R.; Henry, M.; Borentain, P.; Colson, P.; Botta, D.; Tamalet, C. Fulminant hepatitis B associated with a specific insertion in the basal core promoter region of hepatitis B virus DNA after immunosuppressive treatment. Clin. Infect. Dis. 2005, 40, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Buckwold, V.E.; Hon, M.W.; Ou, J.H. Mechanism of suppression of hepatitis B virus precore RNA transcription by a frequent double mutation. J. Virol. 1999, 73, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Akahane, Y.; Hino, K.; Ohta, Y.; Mishiro, S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: Comparative analysis of 40 full-length isolates. Arch. Virol. 1998, 143, 2313–2326. [Google Scholar] [CrossRef]
- Ito, K.; Tanaka, Y.; Kato, M.; Fujiwara, K.; Sugauchi, F.; Sakamoto, T.; Shinkai, N.; Orito, E.; Mizokami, M. Comparison of complete sequences of hepatitis B virus genotype C between inactive carriers and hepatocellular carcinoma patients before and after seroconversion. J. Gastroenterol. 2007, 42, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, N.; Tanaka, Y.; Ito, K.; Mukaide, M.; Hasegawa, I.; Asahina, Y.; Izumi, N.; Yatsuhashi, H.; Orito, E.; Joh, T.; et al. Influence of hepatitis B virus X and core promoter mutations on hepatocellular carcinoma among patients infected with subgenotype C2. J. Clin. Microbiol. 2007, 45, 3191–3197. [Google Scholar] [CrossRef]
- Yuen, M.F.; Tanaka, Y.; Shinkai, N.; Poon, R.T.; But, D.Y.; Fong, D.Y.; Fung, J.; Wong, D.K.; Yuen, J.C.; Mizokami, M.; et al. Risk for hepatocellular carcinoma with respect to hepatitis B virus genotypes B/C, specific mutations of enhancer II/core promoter/precore regions and HBV DNA levels. Gut 2008, 57, 98–102. [Google Scholar] [CrossRef]
- Kim, J.K.; Chang, H.Y.; Lee, J.M.; Baatarkhuu, O.; Yoon, Y.J.; Park, J.Y.; Kim, D.Y.; Han, K.H.; Chon, C.Y.; Ahn, S.H. Specific mutations in the enhancer II/core promoter/precore regions of hepatitis B virus subgenotype C2 in Korean patients with hepatocellular carcinoma. J. Med. Virol. 2009, 81, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhu, Y.; Jin, Y.; Guo, X.; Qian, G.; Chen, T.; Zhang, J.; Wang, J.; Groopman, J.D.; Gu, J.; et al. Temporal acquisition of sequential mutations in the enhancer II and basal core promoter of HBV in individuals at high risk for hepatocellular carcinoma. Carcinogenesis 2011, 32, 63–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lyu, H.; Lee, D.; Chung, Y.H.; Kim, J.A.; Lee, J.H.; Jin, Y.J.; Park, W.; Mathews, P.; Jaffee, E.; Zheng, L.; et al. Synergistic effects of A1896, T1653 and T1762/A1764 mutations in genotype c2 hepatitis B virus on development of hepatocellular carcinoma. J. Viral. Hepat. 2013, 20, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, L.I.; Chen, W. HBV X gene point mutations are associated with the risk of hepatocellular carcinoma: A systematic review and meta-analysis. Mol. Clin. Oncol. 2016, 4, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Kim, K.H.; Lok, A.S.; Tong, S. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J. Virol. 2009, 83, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.; Kremsdorf, D.; Capel, F.; Housset, C.; Dauguet, C.; Petit, M.A.; Brechot, C. Emergence of and takeover by hepatitis B virus (HBV) with rearrangements in the pre-S/S and pre-C/C genes during chronic HBV infection. J. Virol. 1991, 65, 3566–3574. [Google Scholar] [CrossRef]
- Stuyver, L.; De Gendt, S.; Van Geyt, C.; Zoulim, F.; Fried, M.; Schinazi, R.F.; Rossau, R. A new genotype of hepatitis B virus: Complete genome and phylogenetic relatedness. J. Gen. Virol. 2000, 81, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Orito, E.; Gish, R.G.; Sugauchi, F.; Suzuki, S.; Ueda, R.; Miyakawa, Y.; Mizokami, M. Characteristics of hepatitis B virus isolates of genotype G and their phylogenetic differences from the other six genotypes (A through F). J. Virol. 2002, 76, 6131–6137. [Google Scholar] [CrossRef]
- Takahashi, K.; Mishiro, S.; Prince, A.M. Novel hepatitis B virus strain from a chimpanzee of Central Africa (Pan troglodytes troglodytes) with an unusual antigenicity of the core protein. Intervirology 2001, 44, 321–326. [Google Scholar] [CrossRef]
- Utsumi, T.; Wahyuni, R.M.; Lusida, M.I.; Yano, Y.; Priambada, N.P.; Amin, M.; Purwono, P.B.; Istimagfiroh, A.; Soetjipto; Brule, A.; et al. Full genome characterization and phylogenetic analysis of hepatitis B virus in gibbons and a caretaker in Central Kalimantan, Indonesia. Arch. Virol. 2015, 160, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Matsuura, K.; Matsunami, K.; Iio, E.; Nagura, Y.; Nojiri, S.; Kataoka, H. Novel Genetic Rearrangements Termed “Structural Variation Polymorphisms” Contribute to the Genetic Diversity of Orthohepadnaviruses. Viruses 2019, 11, 871. [Google Scholar] [CrossRef]
- Li, K.; Zoulim, F.; Pichoud, C.; Kwei, K.; Villet, S.; Wands, J.; Li, J.; Tong, S. Critical role of the 36-nucleotide insertion in hepatitis B virus genotype G in core protein expression, genome replication, and virion secretion. J. Virol. 2007, 81, 9202–9215. [Google Scholar] [CrossRef] [PubMed]
- Murayama, A.; Yamada, N.; Osaki, Y.; Shiina, M.; Aly, H.H.; Iwamoto, M.; Tsukuda, S.; Watashi, K.; Matsuda, M.; Suzuki, R.; et al. N-Terminal PreS1 Sequence Regulates Efficient Infection of Cell-Culture-Generated Hepatitis B Virus. Hepatology 2020. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Yuan, Q.; Zhang, J.; Wu, J.; Jiang, Q.; Wang, Q.; Xia, N.S.; Tong, S. Naturally occurring 5′ preS1 deletions markedly enhance replication and infectivity of HBV genotype B and genotype C. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wen, X.; Wu, Q.; Bender, D.; Carra, G.; Basic, M.; Kubesch, A.; Peiffer, K.H.; Boller, K.; Hildt, E. The N-Terminus Makes the Difference: Impact of Genotype-Specific Disparities in the N-Terminal Part of The Hepatitis B Virus Large Surface Protein on Morphogenesis of Viral and Subviral Particles. Cells 2020, 9, 1898. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patterns | (n = 70) |
|---|---|
| I. Insertion (+), Deletion (+) | 49 (70.0%) |
| Types of Insertion | |
| A. HNF1 binding site | 24 (34.3%) |
| B. Insertion of unknown origin (X-1) | 7 (10.0%) |
| C. Insertion of unknown origin (X-2) | 3 (4.3%) |
| D. Sequence complementary to part of box α in enhancer II | 6 (8.6%) |
| E. Miscellaneous | 9 (12.9%) |
| II. Insertion (+), Deletion (+), Duplication (+) | 6 (8.6%) |
| Types of Insertion | |
| A. HNF1 binding site | 4 (5.7%) |
| B. Insertion of unknown origin (X-1) | 1 (1.4%) |
| C. Insertion of unknown origin (X-2) | 0 (0.0%) |
| D. Sequence complementary to part of box α in enhancer II | 0 (0.0%) |
| E. Miscellaneous | 1 (1.4%) |
| III. Insertion (+), Duplication (+) | 5 (7.1%) |
| Types of Insertion | |
| A. HNF1 binding site | 1 (2.9%) |
| B. Insertion of unknown origin (X-1) | 2 (4.3%) |
| C. Insertion of unknown origin (X-2) | 0 (0.0%) |
| D. Sequence complementary to part of box α in enhancer II | 0 (0.0%) |
| E. Miscellaneous | 0 (0.0%) |
| IV. Deletion (+), Duplication (+) | 4 (5.7%) |
| V. Duplications (+) | 1 (1.4%) |
| VI. Highly complicated (four or more SVs) | 5 (7.1%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiwara, K. Novel Genetic Rearrangements in Hepatitis B Virus: Complex Structural Variations and Structural Variation Polymorphisms. Viruses 2021, 13, 473. https://doi.org/10.3390/v13030473
Fujiwara K. Novel Genetic Rearrangements in Hepatitis B Virus: Complex Structural Variations and Structural Variation Polymorphisms. Viruses. 2021; 13(3):473. https://doi.org/10.3390/v13030473
Chicago/Turabian StyleFujiwara, Kei. 2021. "Novel Genetic Rearrangements in Hepatitis B Virus: Complex Structural Variations and Structural Variation Polymorphisms" Viruses 13, no. 3: 473. https://doi.org/10.3390/v13030473
APA StyleFujiwara, K. (2021). Novel Genetic Rearrangements in Hepatitis B Virus: Complex Structural Variations and Structural Variation Polymorphisms. Viruses, 13(3), 473. https://doi.org/10.3390/v13030473