
Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning




Abstract
1. HIV-1 Infection: Decades after Implementation of Antiretroviral Therapy
1.1. Innate Restriction Factors Are Natural Host Factors That Interfere with HIV-1 Replication
1.2. TRIM5α: From HIV-1 Restriction Factor to Induction of Antiviral Immunity
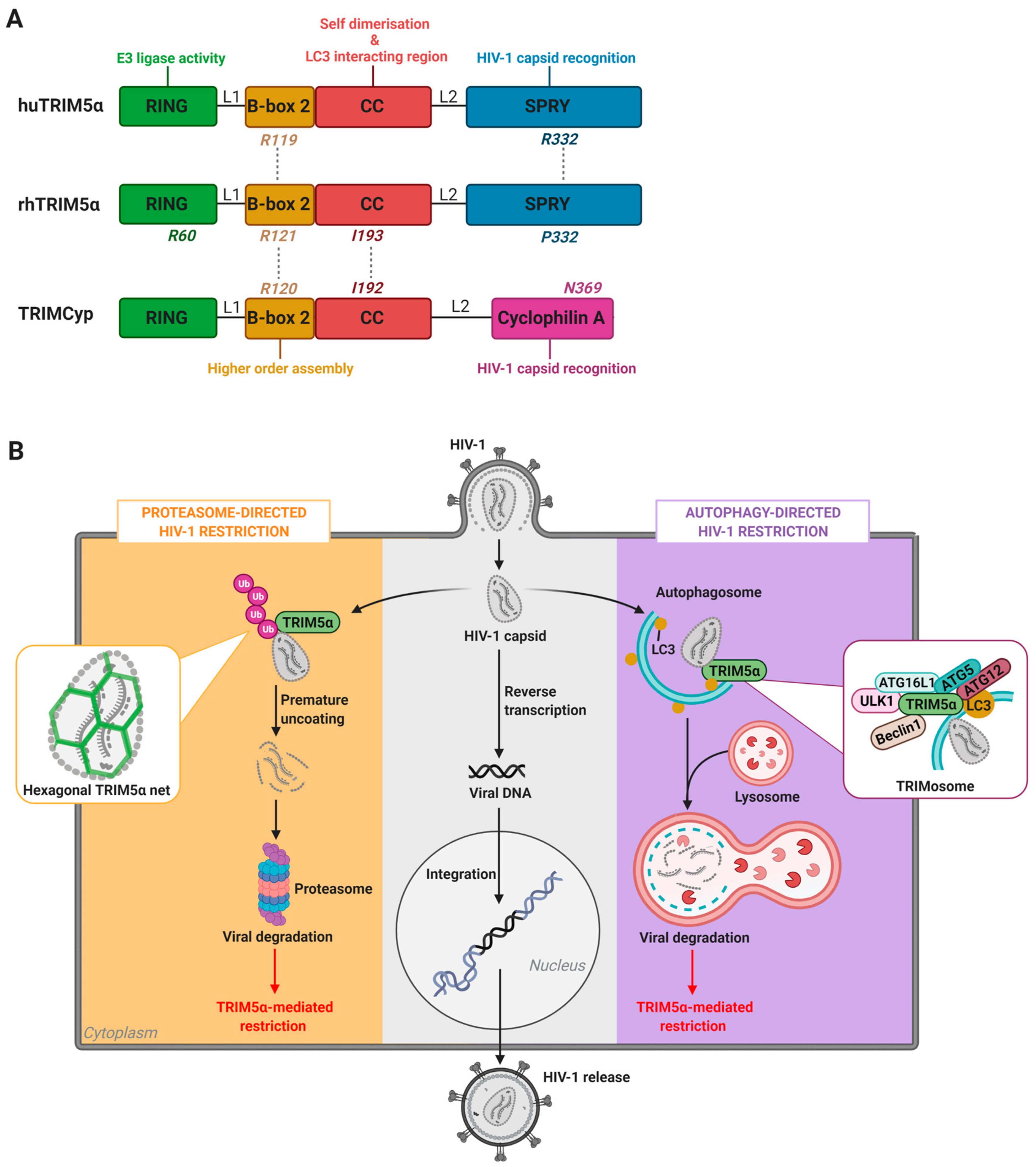
2. TRIM5α-Mediated HIV-1 Restriction Across Species
2.1. TRIM5α of Old World Monkeys: The Case of Rhesus Macaques
2.2. TRIM5α of New World Monkeys: The Case of TRIMCyp
2.3. Human TRIM5α
3. The Nexus Between TRIM5α Antiviral Functions and Host Degradation Pathways
3.1. Cellular Degradation Pathways in HIV-1 Restriction: The Proteasome Versus Autophagy
3.2. TRIM-Mediated Precision Autophagy
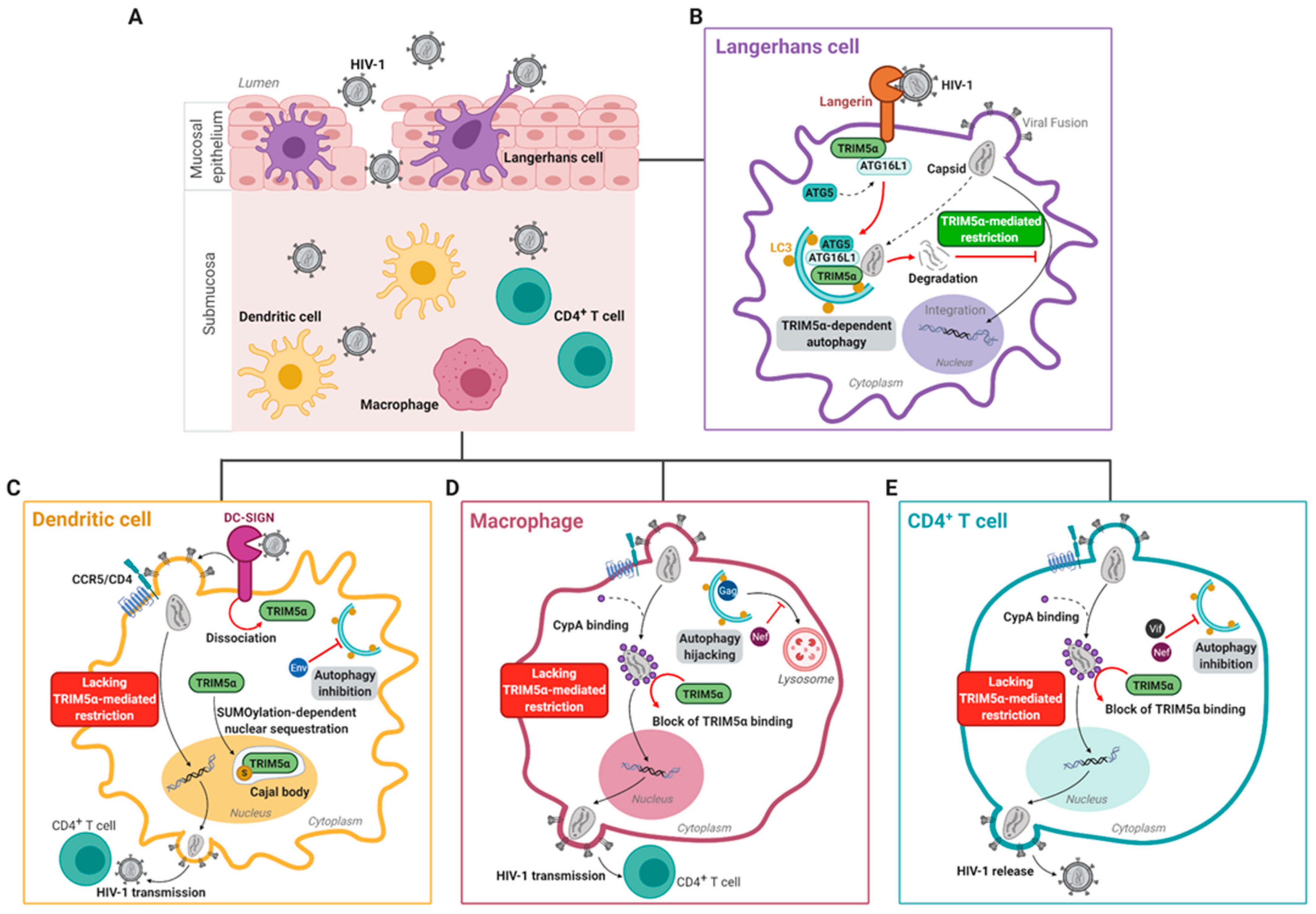
3.3. Human TRIM5α Restricts HIV-1 in Epithelial Langerhans Cells
4. HIV-1 Escapes huTRIM5α-Mediated Restriction in Submucosal DC-SIGN+ Dendritic Cells and CD4+ T Cells
4.1. Submucosal Dendritic Cells
4.2. Macrophages
4.3. T Cells
4.4. Cell-Specific and Autophagy-Dependent Restriction of HIV-1 by Additional Restriction Factors
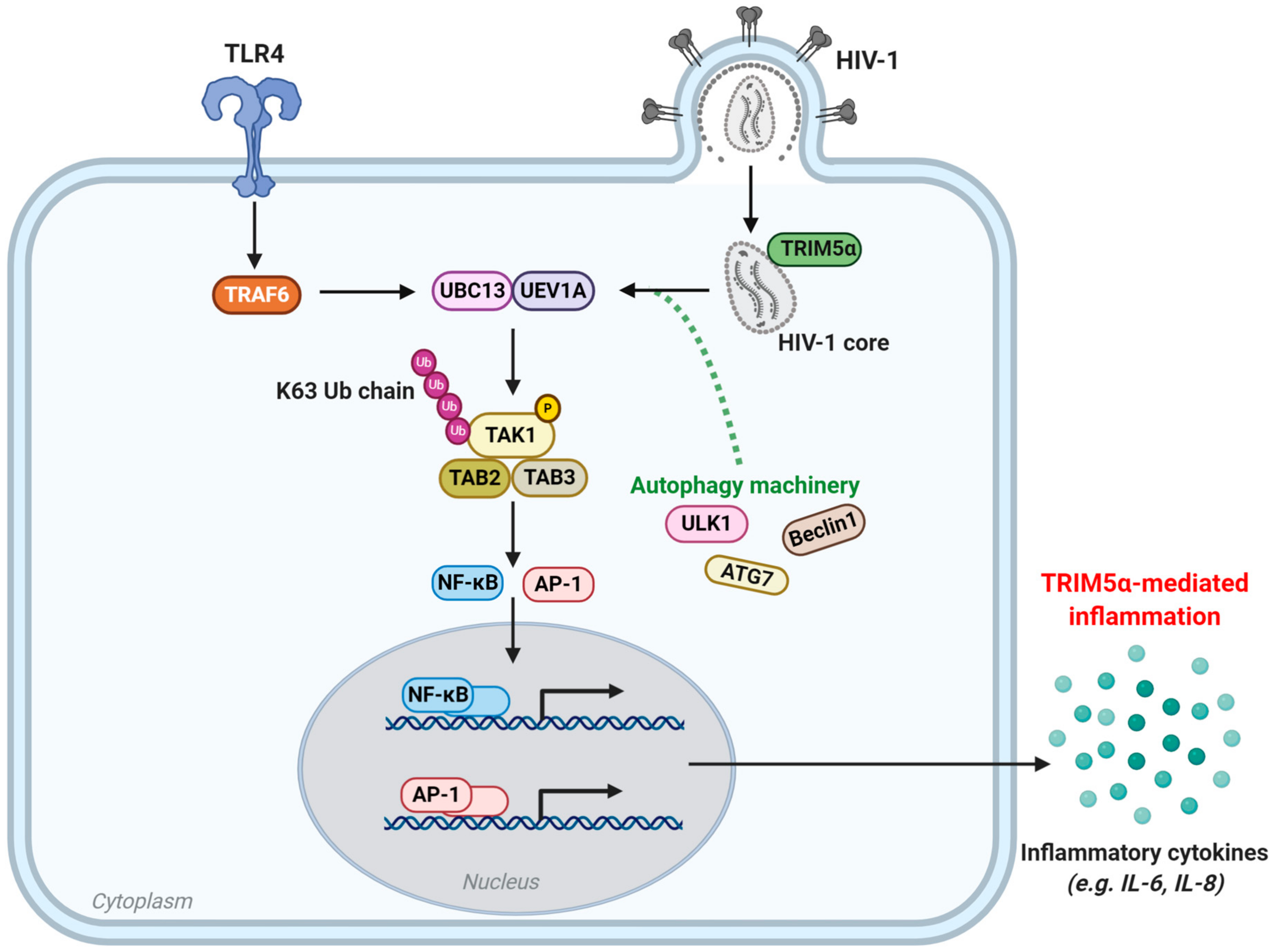
5. TRIM5 as An Innate Immune Sensor
6. In Vivo Relevancy of TRIM5α and Autophagy in HIV-1 Infections
6.1. Impact of huTRIM5α Polymorphisms on Disease Progression
6.2. TRIM5α-Based Therapeutic Strategies for HIV-1
6.3. Impact of Autophagy on HIV-1 Disease Progression and Comorbidities
6.4. Harnessing Autophagy for HIV-1 Management
7. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. HIV/AIDS Factsheets. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 19 October 2020).
- HIV CAUSAL Collaboration; Ray, M.; Logan, R.; Sterne, J.A.C.; Hernández-Díaz, S.; Robins, J.M.; Sabin, C.; Bansi, L.; van Sighem, A.; de Wolf, F.; et al. The effect of combined antiretroviral therapy on the overall mortality of HIV-infected individuals. AIDS 2010, 24, 123–137. [Google Scholar]
- Jean, M.J.; Fiches, G.; Hayashi, T.; Zhu, J.; Fiches, G. Current strategies for elimination of HIV-1 latent reservoirs using chemical compounds targeting host and viral factors. AIDS Res. Hum. Retrovir. 2019, 35, 1–24. [Google Scholar] [CrossRef]
- Hammer, S.M.; Katzenstein, D.A.; Hughes, M.D.; Gundacker, H.; Schooley, R.T.; Haubrich, R.H.; Henry, W.K.; Lederman, M.M.; Phair, J.P.; Niu, M.T.; et al. A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. N. Engl. J. Med. 1996, 335, 1081–1090. [Google Scholar] [CrossRef]
- Egger, M.; Hirschel, B.; Francioli, P.; Sudre, P.; Wirz, M.; Flepp, M.; Rickenbach, M.; Malinverni, R.; Vernazza, P.; Battegay, M. Impact of new antiretroviral combination therapies in HIV infected patients in Switzerland: Prospective multicentre study. Swiss HIV cohort study. BMJ 1997, 315, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.; Chenine, A.-L.; Gruber, A.; Li, P.-L.; Ruprecht, R.M. Long-term productive human immunodeficiency virus infection of CD1a-sorted myeloid dendritic cells. J. Virol. 2005, 79, 602–608. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.N.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.C.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Bruner, K.M.; Hosmane, N.N.; Siliciano, R.F. Towards an HIV-1 cre: Measuring the latent reservoir. Trends Microbiol. 2015, 23, 192–203. [Google Scholar] [CrossRef]
- Mait-Kaufman, J.; Fakioglu, E.; Mesquita, P.M.; Elliott, J.; Lo, Y.; Madan, R.P. Chronic HIV infection is associated with upregulation of proinflammatory cytokine and chemokine and alpha defensin gene expression in colorectal mucosa. AIDS Res. Hum. Retrovir. 2015, 31, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 2011, 53, 1120–1126. [Google Scholar] [CrossRef]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef]
- Kruize, Z.; Kootstra, N.A. The role of macrophages in HIV-1 persistence and pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef] [PubMed]
- Notario-Pérez, F.; Ruiz-Caro, R.; Veiga-Ochoa, M.-D. Historical development of vaginal microbicides to prevent sexual transmission of HIV in women: From past failures to future hopes. Drug Des. Dev. Ther. 2017, 11, 1767–1787. [Google Scholar] [CrossRef]
- McKinnon, L.R.; Liebenberg, L.J.; Yende-Zuma, N.; Archary, D.; Ngcapu, S.; Sivro, A.; Nagelkerke, N.; Lerma, J.G.G.; Kashuba, A.D.; Masson, L.; et al. Genital inflammation undermines the effectiveness of tenofovir gel in preventing HIV acquisition in women. Nat. Med. 2018, 24, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Cheu, R.; Birse, K.; Zevin, A.S.; Perner, M.; Noël-Romas, L.; Grobler, A.; Westmacott, G.; Xie, I.Y.; Butler, J.; et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 2017, 356, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Towers, G.J.; Qasim, W. Gene therapy strategies to exploit TRIM derived restriction factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-De-Castillia, C.; Grütter, C.; Martinetti, G.; Mazzucchelli, L.; Grütter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Investig. 2009, 119, 3035–3047. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction factors: From intrinsic viral restriction to shaping cellular immunity against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Tada, T.; Zhang, Y.; Koyama, T.; Tobiume, M.; Tsunetsugu-Yokota, Y.; Yamaoka, S.; Fujita, H.; Tokunaga, K. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 2015, 21, 1502–1507. [Google Scholar] [CrossRef]
- O’Connor, C.; Pertel, T.; Gray, S.; Robia, S.L.; Bakowska, J.C.; Luban, J.; Campbell, E.M. p62/Sequestosome-1 associates with and sustains the expression of retroviral restriction factor TRIM5α. J. Virol. 2010, 84, 5997–6006. [Google Scholar] [CrossRef]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Kräusslich, H.-G. The molecular architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Genet. 2012, 10, 279–290. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef]
- Lu, J.; Pan, Q.; Rong, L.; Liu, S.-L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Harris, R.S. Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol. 2009, 4, 605–619. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Wu, L. SAMHD1: A new contributor to HIV-1 restriction in resting CD4+T-cells. Retrovirology 2012, 9, 1–5. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Malim, M.H. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; White, T.E.; Schulte, B.; Vieira, D.A.D.S.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nuñez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef]
- Hotter, D.; Sauter, D.; Kirchhoff, F. Guanylate binding protein 5: Impairing virion infectivity by targeting retroviral envelope glycoproteins. Small GTPases 2017, 8, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Krapp, C.; Hotter, D.; Gawanbacht, A.; McLaren, P.J.; Kluge, S.F.; Stürzel, C.M.; Mack, K.; Reith, E.; Engelhart, S.; Ciuffi, A.; et al. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 2016, 19, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, J.; Liu, X. MARCH2 is upregulated in HIV-1 infection and inhibits HIV-1 production through envelope protein translocation or degradation. Virology 2018, 518, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.S.; Johnson, M.C.; Stephens, E.B.; Guatelli, J.C. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral VPU protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Casartelli, N.; Guivel-Benhassine, F.; Bouziat, R.; Brandler, S.; Schwartz, O.; Moris, A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J. Exp. Med. 2010, 207, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Galão, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J.D. Innate sensing of HIV-1 assembly by tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef]
- Ayinde, D.; Bruel, T.; Cardinaud, S.; Porrot, F.; Prado, J.G.; Moris, A.; Schwartz, O. SAMHD1 limits HIV-1 antigen presentation by monocyte-derived dendritic cells. J. Virol. 2015, 89, 6994–7006. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Barrett, B.S.; Guo, K.; Kassiotis, G.; Hasenkrug, K.J.; Dittmer, U.; Gibbert, K.; Santiago, M.L. Tetherin/BST-2 promotes dendritic cell activation and function during acute retrovirus infection. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.K.H.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.; Schubert, D.; LaBonte, J.; Munson, L.; Gibson, S.; Scammell, J.; Ferrigno, P.; Sodroski, J. Species-Specific, Postentry Barriers to Primate Immunodeficiency Virus Infection. J. Virol. 1999, 73, 10020–10028. [Google Scholar] [CrossRef] [PubMed]
- Himathongkham, S.; Luciw, P.A. Restriction of HIV-1 (subtype B) replication at the entry step in rhesus macaque cells. Virology 1996, 219, 485–488. [Google Scholar] [CrossRef]
- Shibata, R.; Sakai, H.; Kawamura, M.; Tokunaga, K.; Adachi, A. Early replication block of human immunodeficiency virus type 1 in monkey cells. J. Gen. Virol. 1995, 76, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Owens, C.M.; Yang, P.C.; Göttlinger, H.; Sodroski, J. Human and simian immunodeficiency virus capsid proteins are major viral determinants of early, postentry replication blocks in simian cells. J. Virol. 2003, 77, 726–731. [Google Scholar] [CrossRef]
- Münk, C.; Brandt, S.M.; Lucero, G.; Landau, N.R. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 13843–13848. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef]
- Danielson, C.M.; Cianci, G.C.; Hope, T.J. Recruitment and dynamics of proteasome association with rhTRIM5α cytoplasmic complexes during HIV-1 infection. Traffic 2012, 13, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Griffero, F.; Qin, X.-R.; Hayashi, F.; Kigawa, T.; Finzi, A.; Sarnak, Z.; Lienlaf, M.; Yokoyama, S.; Sodroski, J. A B-box 2 surface patch important for TRIM5α self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 2009, 83, 10737–10751. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sodroski, J. The TRIM5α B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J. Virol. 2008, 82, 11495–11502. [Google Scholar] [CrossRef]
- Li, Y.-L.; Chandrasekaran, V.; Carter, S.D.; Woodward, C.L.; Christensen, D.E.; Dryden, K.A.; Pornillos, O.; Yeager, M.; Ganser-Pornillos, B.K.; Jensen, G.J.; et al. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. eLife 2016, 5, e16269. [Google Scholar] [CrossRef]
- Carthagena, L.; Parise, M.C.; Ringeard, M.; Chelbi-Alix, M.K.; Hazan, U.; Nisole, S. Implication of TRIM alpha and TRIMCyp in interferon-induced anti-retroviral restriction activities. Retrovirology 2008, 5, 59. [Google Scholar] [CrossRef]
- Portilho, D.M.; Fernandez, J.; Ringeard, M.; Machado, A.K.; Boulay, A.; Mayer, M.; Mullertrutwin, M.C.; Beignon, A.-S.; Kirchhoff, F.; Nisole, S.; et al. Endogenous TRIM5α function is regulated by SUMOylation and nuclear sequestration for efficient innate sensing in dendritic cells. Cell Rep. 2016, 14, 355–369. [Google Scholar] [CrossRef]
- Asaoka, K.; Ikeda, K.; Hishinuma, T.; Horie-Inoue, K.; Takeda, S.; Inoue, S. A retrovirus restriction factor TRIM5α is transcriptionally regulated by interferons. Biochem. Biophys. Res. Commun. 2005, 338, 1950–1956. [Google Scholar] [CrossRef]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; Van Hamme, J.L.; Tigchelaar, W.; Van Der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Jimenez-Guardeño, J.M.; Apolonia, L.; Betancor, G.; Malim, M.H. Immunoproteasome activation enables human TRIM5α restriction of HIV-1. Nat. Microbiol. 2019, 4, 933–940. [Google Scholar] [CrossRef]
- Selyutina, A.; Persaud, M.; Simons, L.M.; Bulnes-Ramos, A.; Buffone, C.; Martinez-Lopez, A.; Scoca, V.; Di Nunzio, F.; Hiatt, J.; Marson, A.; et al. Cyclophilin A prevents HIV-1 restriction in lymphocytes by blocking human TRIM5α binding to the viral core. Cell Rep. 2020, 30, 3766–3777.e6. [Google Scholar] [CrossRef]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Genet. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Welikala, S.; Sodroski, J. Species-specific variation in the B30.2(SPRY) domain of TRIM5α determines the potency of human immunodeficiency virus restriction. J. Virol. 2005, 79, 3139–3145. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Hatziioannou, T.; Yang, A.; Cowan, S.; Bieniasz, P.D. Human tripartite motif 5α domains responsible for retrovirus restriction activity and specificity. J. Virol. 2005, 79, 8969–8978. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Griffero, F.; Kar, A.; Perron, M.; Xiang, S.-H.; Javanbakht, H.; Li, X.; Sodroski, J. Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5α B-Box 2 domain. J. Virol. 2007, 81, 10362–10378. [Google Scholar] [CrossRef] [PubMed]
- Roganowicz, M.D.; Komurlu, S.; Mukherjee, S.; Plewka, J.; Alam, S.L.; Skorupka, K.A.; Wan, Y.; Dawidowski, D.; Cafiso, D.S.; Ganser-Pornillos, B.K.; et al. TRIM5α SPRY/coiled-coil interactions optimize avid retroviral capsid recognition. PLoS Pathog. 2017, 13, e1006686. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5 protein. Proc. Natl. Acad. Sci. USA 2010, 108, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Skorupka, K.A.; Pak, A.J.; Ganser-Pornillos, B.K.; Pornillos, O.; Voth, G.A. TRIM5α self-assembly and compartmentalization of the HIV-1 viral capsid. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Black, L.R.; Aiken, C. TRIM5α Disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5 restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; Vieira, D.A.D.S.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. RING dimerization links higher-order assembly of TRIM5α to synthesis of K63-linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef]
- Lienlaf, M.; Hayashi, F.; Di Nunzio, F.; Tochio, N.; Kigawa, T.; Yokoyama, S.; Diaz-Griffero, F. Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5 rh: Structure of the RING domain of TRIM. J. Virol. 2011, 85, 8725–8737. [Google Scholar] [CrossRef]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome inhibitors uncouple rhesus TRIM5 restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7465–7470. [Google Scholar] [CrossRef]
- Rold, C.J.; Aiken, C. Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Vaysburd, M.; Maslen, S.; Zeng, J.; Skehel, J.M.; Towers, G.J.; James, L.C. Trivalent RING assembly on retroviral capsids activates TRIM5 ubiquitination and innate immune signaling. Cell Host Microbe 2018, 24, 761–775.e6. [Google Scholar] [CrossRef]
- Javanbakht, H.; Diaz-Griffero, F.; Stremlau, M.; Si, Z.; Sodroski, J. The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5α. J. Biol. Chem. 2005, 280, 26933–26940. [Google Scholar] [CrossRef] [PubMed]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV 1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Yap, M.W.; Robertson, L.E.; Haire, L.F.; Taylor, W.R.; Katzourakis, A.; Stoye, J.P.; Taylor, I.A. Structural and functional analysis of prehistoric lentiviruses uncovers an ancient molecular interface. Cell Host Microbe 2010, 8, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Schaller, T.; Eddaoudi, A.; Zhan, H.; Tan, C.P.; Jacobsen, M.; Thrasher, A.J.; Towers, G.J.; Qasim, W. Lentiviral gene therapy against human immunodeficiency virus type 1, using a novel human TRIM21-cyclophilin A restriction factor. Hum. Gene Ther. 2012, 23, 1176–1185. [Google Scholar] [CrossRef]
- Newman, R.M.; Hall, L.; Kirmaier, A.; Pozzi, L.-A.; Pery, E.; Farzan, M.; O’Neil, S.P.; Johnson, W. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog. 2008, 4, e1000003. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Liang, Z.; Qiao, W.; Cen, S.; Liang, C. The highly polymorphic cyclophilin A-binding loop in HIV-1 capsid modulates viral resistance to MxB. Retrovirology 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef]
- Javanbakht, H.; Diaz-Griffero, F.; Yuan, W.; Yeung, D.F.; Li, X.; Song, B.; Sodroski, J. The ability of multimerized cyclophilin A to restrict retrovirus infection. Virology 2007, 367, 19–29. [Google Scholar] [CrossRef]
- Yoo, S.; Myszka, D.G.; Yeh, C.-Y.; McMurray, M.; Hill, C.P.; Sundquist, W.I. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef]
- Wagner, J.M.; Christensen, D.E.; Bhattacharya, A.; Dawidziak, D.M.; Roganowicz, M.D.; Wan, Y.; Pumroy, R.A.; Demeler, B.; Ivanov, D.N.; Ganser-Pornillos, B.K.; et al. A general model for retroviral capsid pattern recognition by TRIM5 proteins. J. Virol. 2018, 92, e01563-17. [Google Scholar] [CrossRef]
- Price, A.J.; Marzetta, F.; Lammers, M.; Ylinen, L.M.J.; Schaller, T.; Wilson, S.J.; Towers, G.J.; James, L.C. Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat. Struct. Mol. Biol. 2009, 16, 1036–1042. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Genet. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Perron, M.J.; Stremlau, M.; Song, B.; Ulm, W.; Mulligan, R.C.; Sodorski, J. TRIM5α mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Wu, L.I.; Akey, J.M.; Emerman, M.; Malik, H.S. High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5α in humans. Curr. Biol. 2006, 16, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, S.; Yap, M.W.; Sheldon, T.; Stoye, J.P. All three variable regions of the TRIM5α B30.2 domain can contribute to the specificity of retrovirus restriction. J. Virol. 2006, 80, 8554–8565. [Google Scholar] [CrossRef] [PubMed]
- Pham, Q.T.; Bouchard, A.; Grütter, M.G.; Berthoux, L. Generation of human TRIM5α mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.W.; Guo, L.; Xin, F.; Yang, X.; Riley, J.L. Stabilized human TRIM5α protects human T cells from HIV-1 infection. Mol. Ther. 2014, 22, 1084–1095. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of arginine 332 allows human TRIM5α to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. eLife 2018, 7. [Google Scholar] [CrossRef]
- Lu, K.; den Brave, F.; Jentsch, S. Receptor oligomerization guides pathway choice between proteasomal and autophagic degradation. Nat. Cell Biol. 2017, 19, 732–739. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2012, 20, 21–30. [Google Scholar] [CrossRef]
- Rajalingam, K.; Dikic, I. Expanding the ubiquitin code. Cell 2016, 164, 1074–1074.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Elmira, E.; Rohondia, S.; Wang, J.; Liu, J.; Dou, Q.P. A patent review of the ubiquitin ligase system: 2015–2018. Expert Opin. Ther. Patents 2018, 28, 919–937. [Google Scholar] [CrossRef]
- Seissler, T.; Marquet, R.; Paillart, J.C. Hijacking of the ubiquitin/proteasome pathway by the hiv auxiliary proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 47–60. [Google Scholar] [CrossRef]
- Tai, H.-C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Vinod, V.; Simons, J.D.; Boudina, S. Protein and mitochondria quality control mechanisms and cardiac aging. Cells 2020, 9, 933. [Google Scholar] [CrossRef]
- Rojas, V.K.; Park, I.-W. Role of the ubiquitin proteasome system (UPS) in the HIV-1 life cycle. Int. J. Mol. Sci. 2019, 20, 2984. [Google Scholar] [CrossRef] [PubMed]
- Lennemann, N.J.; Coyne, C.B. Catch me if you can: The link between autophagy and viruses. PLoS Pathog. 2015, 11, e1004685. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Agrotis, A.; Ketteler, R. On ATG4B as drug target for treatment of solid tumours—The knowns and the unknowns. Cells 2019, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Noda, N.N.; Fujioka, Y.; Hanada, T.; Ohsumi, Y.; Inagaki, F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013, 14, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jia, J.; Claude-Taupin, A.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; et al. Cellular and molecular mechanism for secretory autophagy. Autophagy 2017, 13, 1084–1085. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 2014, 10, 1327–1334. [Google Scholar] [CrossRef]
- Svenning, S.; Johansen, T. Selective autophagy. Essays Biochem. 2013, 55, 79–92. [Google Scholar] [CrossRef]
- Szalai, P.; Hagen, L.K.; Sætre, F.; Luhr, M.; Sponheim, M.; Øverbye, A.; Mills, I.G.; Seglen, P.O.; Engedal, N. Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 2015, 333, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Keown, J.R.; Black, M.M.; Ferron, A.; Yap, M.W.; Barnett, M.J.; Pearce, F.G.; Stoye, J.P.; Goldstone, D.C. A helical LC3-interacting region mediates the interaction between the retroviral restriction factor Trim5α and mammalian autophagy-related ATG8 proteins. J. Biol. Chem. 2018, 293, 18378–18386. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Mandell, M.; Deretic, V. Precision autophagy directed by receptor regulators—Emerging examples within the TRIM family. J. Cell Sci. 2016, 129, 881–891. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Christensen, D.E.; Nelson, C.; Tan, C.P.; Schaller, T.; Lehner, P.J.; Sundquist, W.I.; Towers, G.J. TRIM 5α requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J. 2015, 34, 2078–2095. [Google Scholar] [CrossRef]
- Yamauchi, K.; Wada, K.; Tanji, K.; Tanaka, M.; Kamitani, T. Ubiquitination of E3 ubiquitin ligase TRIM5α and its potential role. FEBS J. 2008, 275, 1540–1555. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Hatziioannou, T.; Zhang, F.; Cowan, S.; Bieniasz, P.D. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 2005, 79, 15567–15572. [Google Scholar] [CrossRef]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Münch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef]
- Lebovitz, C.B.; DeVorkin, L.; Bosc, D.G.; Rothe, K.; Singh, J.; Bally, M.B.; Jiang, X.; Young, R.N.; Lum, J.J.; Gorski, S.M. Precision autophagy: Will the next wave of selective autophagy markers and specific autophagy inhibitors feed clinical pipelines? Autophagy 2015, 11, 1949–1952. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM proteins in autophagy: Selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020, 27, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef]
- Imam, S.; Talley, S.; Nelson, R.S.; Dharan, A.; O’Connor, C.; Hope, T.J.; Campbell, E.M. TRIM5α degradation via autophagy is not required for retroviral restriction. J. Virol. 2016, 90, 3400–3410. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Mamede, J.I.; Hope, T.J.; Jensen, G.J. Correlated cryogenic fluorescence microscopy and electron cryo-tomography shows that exogenous TRIM5α can form hexagonal lattices or autophagy aggregates in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 29702–29711. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- De Witte, L.; Nabatov, A.; Pion, M.; Fluitsma, D.; De Jong, M.A.W.P.; De Gruijl, T.; Piguet, V.; Van Kooyk, Y.; Geijtenbeek, T.B.H. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat. Med. 2007, 13, 367–371. [Google Scholar] [CrossRef]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Geijtenbeek, T.B.H. HIV-1 border patrols: Langerhans cells control antiviral responses and viral transmission. Future Virol. 2015, 10, 1231–1243. [Google Scholar] [CrossRef]
- Hladik, F.; Sakchalathorn, P.; Ballweber, L.; Lentz, G.; Fialkow, M.; Eschenbach, D.; McElrath, M.J. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunology 2007, 26, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Qualbani, M.; Thomas, E.K.; Orenstein, J.M.; Blauvelt, A. Low levels of productive HIV infection in Langerhans cell-like dendritic cells differentiated in the presence of TGF-β1 and increased viral replication with CD40 ligand-induced maturation. Eur. J. Immunol. 2001, 31, 360–368. [Google Scholar] [CrossRef]
- Valladeau, J.; Ravel, O.; Dezutter-Dambuyant, C.; Moore, K.; Kleijmeer, M.; Liu, Y.; Duvert-Frances, V.; Vincent, C.; Schmitt, D.; Davoust, J.; et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 2000, 12, 71–81. [Google Scholar] [CrossRef]
- Van Der Vlist, M.; Geijtenbeek, T.B.H. Langerin functions as an antiviral receptor on Langerhans cells. Immunol. Cell Biol. 2010, 88, 410–415. [Google Scholar] [CrossRef]
- Berg, L.M.V.D.; Ribeiro, C.M.S.; Zijlstra-Willems, E.M.; De Witte, L.; Fluitsma, D.; Tigchelaar, W.; Everts, V.; Geijtenbeek, T.B.H. Caveolin-1 mediated uptake via langerin restricts HIV-1 infection in human Langerhans cells. Retrovirology 2014, 11, 1–9. [Google Scholar] [CrossRef]
- Czubala, M.A.; Finsterbusch, K.; Ivory, M.O.; Mitchell, J.P.; Ahmed, Z.; Shimauchi, T.; Karoo, R.O.; Coulman, S.A.; Gateley, C.; Birchall, J.C.; et al. TGFβ Induces a SAMHD1-independent post-entry restriction to HIV-1 infection of human epithelial langerhans cells. J. Investig. Dermatol. 2016, 136, 1981–1989. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hertoghs, N.; Geijtenbeek, T.B.H.; Ribeiro, C.M.S. Interplay between HIV-1 innate sensing and restriction in mucosal dendritic cells: Balancing defense and viral transmission. Curr. Opin. Virol. 2017, 22, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Sakoh-Nakatogawa, M.; Matoba, K.; Asai, E.; Kirisako, H.; Ishii, J.; Noda, N.N.; Inagaki, F.; Nakatogawa, H.; Ohsumi, Y. Atg12–Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat. Struct. Mol. Biol. 2013, 20, 433–439. [Google Scholar] [CrossRef]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef]
- Gramberg, T.; Soilleux, E.; Fisch, T.; Lalor, P.F.; Hofmann, H.; Wheeldon, S.; Cotterill, A.; Wegele, A.; Winkler, T.; Adams, D.H.; et al. Interactions of LSECtin and DC-SIGN/DC-SIGNR with viral ligands: Differential pH dependence, internalization and virion binding. Virology 2008, 373, 189–201. [Google Scholar] [CrossRef]
- Trifonova, R.T.; Bollman, B.; Barteneva, N.S.; Lieberman, J. Myeloid cells in intact human cervical explants capture HIV and can transmit it to CD4 T cells. Front. Immunol. 2018, 9, 2719. [Google Scholar] [CrossRef]
- Bertram, K.M.; Botting, R.A.; Baharlou, H.; Rhodes, J.W.; Rana, H.; Graham, J.D.; Patrick, E.; Fletcher, J.; Plasto, T.M.; Truong, N.R.; et al. Identification of HIV transmitting CD11c+ human epidermal dendritic cells. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- McIlroy, D.; Autran, B.; Cheynier, R.; Wain-Hobson, S.; Clauvel, J.P.; Oksenhendler, E.; Debré, P.; Hosmalin, A. Infection frequency of dendritic cells and CD4+ T lymphocytes in spleens of human immunodeficiency virus-positive patients. J. Virol. 1995, 69, 4737–4745. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Van Der Vlist, M.; Berg, L.M.V.D.; Dunnen, J.D.; Litjens, M.; Geijtenbeek, T.B.H. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; Van Vliet, S.J.; Van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Hertoghs, N.; Nijmeijer, B.M.; Van Teijlingen, N.H.; Fenton-May, A.E.; Kaptein, T.M.; Van Hamme, J.L.; Kappes, J.C.; Kootstra, N.A.; Hahn, B.H.; Borrow, P.; et al. Sexually transmitted founder HIV-1 viruses are relatively resistant to Langerhans cell-mediated restriction. PLoS ONE 2019, 14, e0226651. [Google Scholar] [CrossRef]
- De Jong, M.A.; de Witte, L.; Oudhoff, M.J.; Gringhuis, S.I.; Gallay, P.; Geijtenbeek, T.B. TNF-α and TLR agonists increase susceptibility to HIV-1 transmission by human Langerhans cells ex vivo. J. Clin. Investig. 2008, 118, 3440–3452. [Google Scholar] [CrossRef]
- Hester, R.A.; Kennedy, S.B. Candida infection as a risk factor for HIV transmission. J. Women’s Health 2003, 12, 487–494. [Google Scholar] [CrossRef]
- Fleming, D.T.; Wasserheit, J.N. From epidemiological synergy to public health policy and practice: The contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex. Transm. Infect. 1999, 75, 3–17. [Google Scholar] [CrossRef]
- Engering, A.; Geijtenbeek, T.B.; van Vliet, S.J.; Wijers, M.; van Liempt, E.; Demaurex, N.; Lanzavecchia, A.; Fransen, J.; Figdor, C.G.; Piguet, V.; et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for fresentation to T cells. J. Immunol. 2002, 168, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science 1992, 257, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.; Betjes, M.; Romani, N.; Hirmand, H.; Cameron, P.; Hoffman, L.; Gezelter, S.; Schuler, G.; Steinman, R. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell 1994, 78, 389–398. [Google Scholar] [CrossRef]
- Smed-Sörensen, A.; Loré, K.; Vasudevan, J.; Louder, M.K.; Andersson, J.; Mascola, J.R.; Spetz, A.-L.; Koup, R.A. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J. Virol. 2005, 79, 8861–8869. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van Het Hof, B.; van Kooyk, Y.; Geijtenbeek, T.B. C-type lectin DC-SIGN modulates toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-κB. Immunity 2007, 26, 605–616. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Dunnen, J.D.; Litjens, M.; Van Der Vlist, M.; Geijtenbeek, T.B.H. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat. Immunol. 2009, 10, 1081–1088. [Google Scholar] [CrossRef]
- Cloherty, A.P.M.; van Teijlingen, N.H.; Eisden, T.T.H.D.; van Hamme, J.L.; Rader, A.G.; Geijtenbeek, T.B.H.; Schreurs, R.R.C.E.; Ribeiro, C.M.S. Autophagy-enhancing drugs limit mucosal HIV-1 acquisition and suppress viral replication ex vivo. Sci. Rep. 2021, 11, 4767. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Thali, M.; Bukovsky, A.A.; Kondo, E.; Rosenwlrth, B.; Walsh, C.T.; Sodroski, J.; Göttlinger, H.G. Functional association of cyclophilin A with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar] [CrossRef]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef]
- Jin, C.; Li, J.; Cheng, L.; Liu, F.; Wu, N. Gp120 binding with DC-SIGN induces reactivation of HIV-1 provirus via the NF-κB signaling pathway. Acta Biochim. Biophys. Sin. 2015, 48, 275–281. [Google Scholar] [CrossRef]
- Arhel, N.J.; Nisole, S.; Carthagena, L.; Coutant, F.; Souque, P.; Brussel, A.; Estaquier, J.; Charneau, P. Lack of endogenous TRIM5α-mediated restriction in rhesus macaque dendritic cells. Blood 2008, 112, 3772–3776. [Google Scholar] [CrossRef]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An unhealthy constellation. Cell Host Microbe 2016, 19, 304–310. [Google Scholar] [CrossRef]
- Haldar, M.; Murphy, K.M. Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 2014, 262, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Lopez, P.; Koh, W.H.; Hnatiuk, R.; Murooka, T.T. HIV infection stabilizes macrophage-T cell interactions to promote cell-cell HIV spread. J. Virol. 2019, 93, 805–819. [Google Scholar] [CrossRef]
- Carr, J.; Hocking, H.; Li, P.; Burrell, C. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 1999, 265, 319–329. [Google Scholar] [CrossRef]
- Duncan, C.J.A.; Russell, R.A.; Sattentau, Q.J. High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS 2013, 27, 2201–2206. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading TAT in CD4+T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional regulation of autophagy: Mechanisms and diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Young, L.N.; Morris, K.L.; Von Bülow, S.; Schöneberg, J.; Yamamoto-Imoto, H.; Oe, Y.; Yamamoto, K.; Nakamura, S.; Stjepanovic, G.; et al. Bidirectional control of autophagy by BECN1 BARA domain dynamics. Mol. Cell 2019, 73, 339–353.e6. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy 2020, 1–25. [Google Scholar] [CrossRef]
- Veillette, M.; Bichel, K.; Pawlica, P.; Freund, S.M.V.; Plourde, M.B.; Pham, Q.T.; Reyes-Moreno, C.; James, L.C.; Berthoux, L. The V86M mutation in HIV-1 capsid confers resistance to TRIM5α by abrogation of cyclophilin a-dependent restriction and enhancement of viral nuclear import. Retrovirology 2013, 10, 25. [Google Scholar] [CrossRef]
- Trifonova, R.T.; Lieberman, J.; Van Baarle, D. Distribution of immune cells in the human cervix and implications for HIV transmission. Am. J. Reprod. Immunol. 2014, 71, 252–264. [Google Scholar] [CrossRef]
- Borel, S.; Robert-Hebmann, V.; AlFaisal, J.; Jain, A.; Faure, M.; Espert, L.; Chaloin, L.; Paillart, J.-C.; Johansen, T.; Biard-Piechaczyk, M. HIV-1 viral infectivity factor interacts with microtubule-associated protein light chain 3 and inhibits autophagy. AIDS 2015, 29, 275–286. [Google Scholar] [CrossRef]
- Sanfridson, A.; Hester, S.; Doyle, C. Nef proteins encoded by human and simian immunodeficiency viruses induce the accumulation of endosomes and lysosomes in human T cells. Proc. Natl. Acad. Sci. USA 1997, 94, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, B.; Wittmann, S.; Lagisquet, J.; Deutschmann, J.; Eissmann, K.; Ross, J.J.; Biesinger, B.; Gramberg, T. Human TRIM5α senses and restricts LINE-1 elements. Proc. Natl. Acad. Sci. USA 2020, 117, 17965–17976. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.H.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 2012, 18, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Swiderski, M.; Khan, A.; Gitzen, A.; Majadly, A.; Freed, E.O. The viral protein U (VPU)-interacting host protein ATP6V0C down-regulates cell-surface expression of tetherin and thereby contributes to HIV-1 release. J. Biol. Chem. 2020, 295, 7327–7340. [Google Scholar] [CrossRef]
- Blanchet, F.P.; Mitchell, J.P.; Piguet, V. Beta-TrCP dependency of HIV-1 VPU-induced downregulation of CD4 and BST-2/Tetherin. Curr. HIV Res. 2012, 10, 307–314. [Google Scholar] [CrossRef]
- Miyakawa, K.; Ryo, A.; Murakami, T.; Ohba, K.; Yamaoka, S.; Fukuda, M.; Guatelli, J.; Yamamoto, N. BCA2/Rabring7 promotes tetherin-dependent HIV-1 restriction. PLoS Pathog. 2009, 5, e1000700. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Li, W.; Misra, A.; Yue, F.; Song, K.; Chen, Q.; Guo, G.; Yi, J.; Kimata, J.T.; Liu, L. The viral restriction factor tetherin prevents leucine-rich pentatricopeptide repeat-containing protein (LRPPRC) from association with beclin 1 and B-cell CLL/lymphoma 2 (Bcl-2) and enhances autophagy and mitophagy. J. Biol. Chem. 2015, 290, 7269–7279. [Google Scholar] [CrossRef]
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell 2017, 68, 308–322.e4. [Google Scholar] [CrossRef]
- Tareen, S.U.; Emerman, M. Human Trim5α has additional activities that are uncoupled from retroviral capsid recognition. Virology 2011, 409, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Chisholm, D.; Kell, A.M.; Mandell, M.A. A non-canonical role for the autophagy machinery in anti-retroviral signaling mediated by TRIM5α. PLoS Pathog. 2020, 16, e1009017. [Google Scholar] [CrossRef]
- Cheney, K.M.; McKnight, Á. Interferon-alpha mediates restriction of human immunodeficiency virus type-1 replication in primary human macrophages at an early stage of replication. PLoS ONE 2010, 5, e13521. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.D.; Smiley, J.R.; Bushman, F.D. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008, 4, e1000007. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, J.; Zhang, C.; Huang, S.; Nunnari, G.; Wang, F.-X.; Tong, X.; Gao, L.; Nikisher, K.; Zhang, H. Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T Cells. J. Virol. 2006, 80, 7645–7657. [Google Scholar] [CrossRef]
- Iwasaki, A. Innate immune recognition of HIV-1. Immunity 2012, 37, 389–398. [Google Scholar] [CrossRef]
- Lascano, J.; Uchil, P.D.; Mothes, W.; Luban, J. TRIM5 retroviral restriction activity correlates with the ability to induce innate immune signaling. J. Virol. 2015, 90, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, H.; An, P.; Gold, B.; Petersen, D.C.; O’Huigin, C.; Nelson, G.W.; O’Brien, S.J.; Kirk, G.D.; Detels, R.; Buchbinder, S.; et al. Effects of human TRIM5α polymorphisms on antiretroviral function and susceptibility to human immunodeficiency virus infection. Virology 2006, 354, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Price, H.; Lacap, P.; Tuff, J.; Wachihi, C.; Kimani, J.; Ball, T.B.; Luo, M.; Plummer, F.A. A TRIM5α exon 2 polymorphism is associated with protection from HIV-1 infection in the Pumwani sex worker cohort. AIDS 2010, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.C.; Coelho, A.V.C.; Arraes, L.C.; Brandão, L.A.C.; Crovella, S.; Guimarães, R.L. TRIM5 gene polymorphisms in HIV-1-infected patients and healthy controls from Northeastern Brazil. Immunol. Res. 2016, 64, 1237–1242. [Google Scholar] [CrossRef]
- Speelmon, E.C.; Livingston-Rosanoff, D.; Li, S.S.; Vu, Q.; Bui, J.; Geraghty, D.E.; Zhao, L.P.; McElrath, M.J. Genetic association of the antiviral restriction factor TRIM5α with human immunodeficiency virus type 1 infection. J. Virol. 2006, 80, 2463–2471. [Google Scholar] [CrossRef]
- Nakajima, T.; Nakayama, E.E.; Kaur, G.; Terunuma, H.; Mimaya, J.-I.; Ohtani, H.; Mehra, N.; Shioda, T.; Kimura, A. Impact of novel TRIM5α variants, Gly110Arg and G176del, on the anti-HIV-1 activity and the susceptibility to HIV-1 infection. AIDS 2009, 23, 2091–2100. [Google Scholar] [CrossRef]
- Liu, F.-L.; Qiu, Y.-Q.; Li, H.; Kuang, Y.-Q.; Tang, X.; Cao, G.; Tang, N.L.S.; Zheng, Y.-T. An HIV-1 resistance polymorphism in TRIM5α gene among chinese intravenous drug users. J. Acquir. Immune Defic. Syndr. (JAIDS) 2011, 56, 306–311. [Google Scholar] [CrossRef]
- Van Manen, D.; Rits, M.A.N.; Beugeling, C.; Van Dort, K.; Schuitemaker, H.; Kootstra, N.A. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 2008, 4, e18. [Google Scholar] [CrossRef]
- Nakayama, E.E.; Nakajima, T.; Kaur, G.; Mimaya, J.-I.; Terunuma, H.; Mehra, N.; Kimura, A.; Shioda, T. A naturally occurring single amino acid substitution in human TRIM5α linker region affects its anti-HIV type 1 activity and susceptibility to HIV type 1 infection. AIDS Res. Hum. Retrovir. 2013, 29, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Samani, D.; Ghate, M.V.; Gangakhedkar, R.R. Impact of cellular restriction gene (TRIM5α, BST-2) polymorphisms on the acquisition of HIV-1 and disease progression. J. Gene Med. 2018, 20, e3004. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Haralambieva, I.H.; Dhiman, N.; O’Byrne, M.M.; Pankratz, V.S.; Jacobson, R.M.; Poland, G.A. Polymorphisms in the vitamin A receptor and innate immunity genes influence the antibody response to rubella vaccination. J. Infect. Dis. 2010, 201, 207–213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ovsyannikova, I.G.; Haralambieva, I.H.; Vierkant, R.A.; O’Byrne, M.M.; Poland, G.A. Associations between polymorphisms in the antiviral TRIM genes and measles vaccine immunity. Hum. Immunol. 2013, 74, 768–774. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mobasheri, S.; Irani, N.; Sepahi, A.A.; Sakhaee, F.; Jamnani, F.R.; Vaziri, F.; Siadat, S.D.; Fateh, A. Evaluation of TRIM5 and TRIM22 polymorphisms on treatment responses in Iranian patients with chronic hepatitis C virus infection. Gene 2018, 676, 95–100. [Google Scholar] [CrossRef]
- Medrano, L.M.; Rallón, N.; Berenguer, J.; Jiménez-Sousa, M.Á.; Soriano, V.; Aldámiz-Echevarria, T.; Fernández-Rodríguez, A.; García, M.; Tejerina, F.; Martinez, I.; et al. Relationship of TRIM5 and TRIM22 polymorphisms with liver disease and HCV clearance after antiviral therapy in HIV/HCV coinfected patients. J. Transl. Med. 2016, 14, 257. [Google Scholar] [CrossRef]
- Abdel-Mohsen, M.; Wang, C.; Strain, M.C.; Lada, S.M.; Deng, X.; Cockerham, L.R.; Pilcher, C.D.; Hecht, F.M.; Liegler, T.; Richman, D.D.; et al. Select host restriction factors are associated with HIV persistence during antiretroviral therapy. AIDS 2015, 29, 411–420. [Google Scholar] [CrossRef]
- Raposo, R.A.S.; Abdel-Mohsen, M.; Deng, X.; Hecht, F.M.; Pilcher, C.D.; Pillai, S.K.; Nixon, D.F. Dynamic regulation of host restriction factor expression over the course of HIV-1 infection in vivo. J. Virol. 2014, 88, 11624–11629. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Johansen, T.; Deretic, V. TRIM-directed selective autophagy regulates immune activation. Autophagy 2017, 13, 989–990. [Google Scholar] [CrossRef]
- Yap, M.W.; Dodding, M.P.; Stoye, J.P. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J. Virol. 2006, 80, 4061–4067. [Google Scholar] [CrossRef]
- Walker, J.E.; Chen, R.X.; McGee, J.; Nacey, C.; Pollard, R.B.; Abedi, M.; Bauer, G.; Nolta, J.A.; Anderson, J.S. Generation of an HIV-1-resistant immune system with CD34+ hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J. Virol. 2012, 86, 5719–5729. [Google Scholar] [CrossRef]
- Anderson, J.; Akkina, R. Human immunodeficiency virus type 1 restriction by human—Rhesus chimeric tripartite motif 5α (TRIM5α) in CD34+ cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum. Gene Ther. 2008, 19, 217–228. [Google Scholar] [CrossRef]
- Morgan, R.A.; Gray, D.; Lomova, A.; Kohn, D.B. Hematopoietic stem cell gene therapy: Progress and lessons learned. Cell Stem Cell 2017, 21, 574–590. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.-L.; Trout, R.N.; Spector, S.A. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting memory CD4+ T Cells. Cell Host Microbe 2018, 24, 689–702.e7. [Google Scholar] [CrossRef]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Kaminski, R.; Mullen, B.; Gordon, J.; Burdo, T.H.; Cheung, J.Y.; Feldman, A.M.; Madesh, M.; Khalili, K. HIV-1 Nef-induced cardiotoxicity through dysregulation of autophagy. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Remick, J.; Georgiopoulou, V.; Marti, C.; Ofotokun, I.; Kalogeropoulos, A.; Lewis, W.; Butler, J. Heart failure in patients with human immunodeficiency virus infection: Epidemiology, pathophysiology, treatment, and future research. Circulation 2014, 129, 1781–1789. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Shanmughapriya, S.; Ahooyi, T.M.; Knezevic, T.; Gupta, M.K.; Kontos, C.D.; McClung, J.M.; Madesh, M.; Gordon, J.; Feldman, A.M.; et al. Dysregulation of mitochondrial bioenergetics and quality control by HIV-1 Tat in cardiomyocytes. J. Cell. Physiol. 2018, 233, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of autophagy in HIV infection and pathogenesis. J. Intern. Med. 2017, 281, 422–432. [Google Scholar] [CrossRef]
- Brew, B.J.; Chan, P. Update on HIV dementia and HIV-associated neurocognitive disorders. Curr. Neurol. Neurosci. Rep. 2014, 14, 468. [Google Scholar] [CrossRef]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Mehla, R.; Chauhan, A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J. Neuroimmunol. 2015, 285, 106–118. [Google Scholar] [CrossRef]
- Zhang, G.; Luk, B.T.; Hamidy, M.; Zhang, L.; Spector, S.A. Induction of a Na+/K+-ATPase-dependent form of autophagy triggers preferential cell death of human immunodeficiency virus type-1-infected macrophages. Autophagy 2018, 14, 1359–1375. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog. 2012, 8, e1003017. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.N.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]
- Coulon, P.-G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-infected dendritic cells present endogenous MHC class II—Restricted antigens to HIV-specific CD4+ T cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Chemali, M.; Radtke, K.; Desjardins, M.; English, L. Alternative pathways for MHC class I presentation: A new function for autophagy. Cell. Mol. Life Sci. 2011, 68, 1533–1541. [Google Scholar] [CrossRef]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Effect | SNP | Domain | Implication | Outcome | p | Ref. |
|---|---|---|---|---|---|---|
| HIV-1 susceptibility | rs3740996 | RING | Higher frequencies in SN 1 compared to SC 2 | Protective | <0.043 | [200,205] |
| No association with HIV-1 susceptibility | No effect | - | [203] | |||
| rs10838525 | CC | Higher frequencies in HREU 3 compared to SC | Protective | <0.02 | [200,201] | |
| rs904375 | Intron 4 | Higher frequencies in HIV-1 positive individuals compared to healthy controls | Negative | 0.46 | [208] | |
| rs11038628 | Linker 2 | Higher frequencies in HIV-1 positive individuals compared to healthy controls | Negative | 0.026 | [207] | |
| G110R | B-box | Higher frequencies in HIV-1 positive individuals compared to healthy controls | Negative | 0.002 | [204] | |
| Disease progression | rs3740996 | RING | Homozygous genotype resulted in accelerated disease progression | Negative | - | [95,206] |
| rs10838525 | CC | Protective effect observed only when CXCR4-using HIV-1 variants were present and when a viral load of 10 4,5 copies/mL plasma was used as endpoint | Partial effect | 0.008 (HZ 4); 0.03 (HO 5) | [206] | |
| rs3824949 | 5′ UTR | Accelerated disease progression when in combination with the 136Q allele (rs10838525) | Negative | 0.009 | [206] | |
| Innate; adaptive immunity | rs3824949 | 5′ UTR | Improved RVR 6, EVR 7, SVR 8 for Hepatitis C virus | Protective | <0.001 | [211] |
| rs3824949 rs7122620 | 5′ UTR 3′ UTR | Allele dose-related increase in rubella antibody response | Protective | <0.015 | [209] | |
| Differences in measles specific antibody levels. | Protective | <0.02 | [210] | |||
| rs7122620 | 3′ UTR | Increased IL-2 secretion levels upon measles vaccination | Protective | <0.015 | [210] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cloherty, A.P.M.; Rader, A.G.; Compeer, B.; Ribeiro, C.M.S. Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning. Viruses 2021, 13, 320. https://doi.org/10.3390/v13020320
Cloherty APM, Rader AG, Compeer B, Ribeiro CMS. Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning. Viruses. 2021; 13(2):320. https://doi.org/10.3390/v13020320
Chicago/Turabian StyleCloherty, Alexandra P. M., Anusca G. Rader, Brandon Compeer, and Carla M. S. Ribeiro. 2021. "Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning" Viruses 13, no. 2: 320. https://doi.org/10.3390/v13020320
APA StyleCloherty, A. P. M., Rader, A. G., Compeer, B., & Ribeiro, C. M. S. (2021). Human TRIM5α: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning. Viruses, 13(2), 320. https://doi.org/10.3390/v13020320