
Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture

 , , ,
, , ,
Abstract
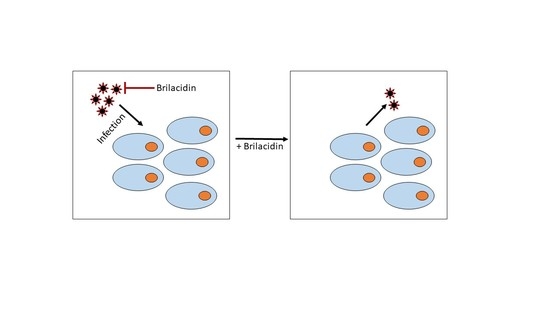
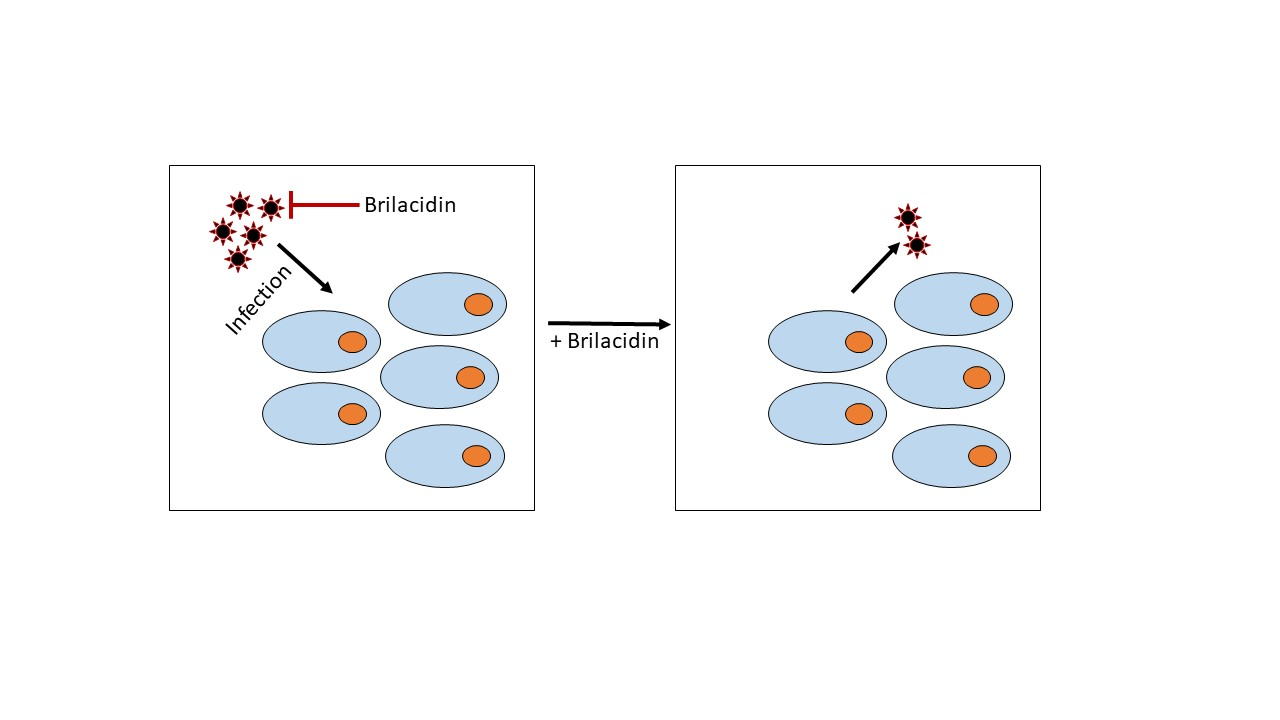{kind=link}
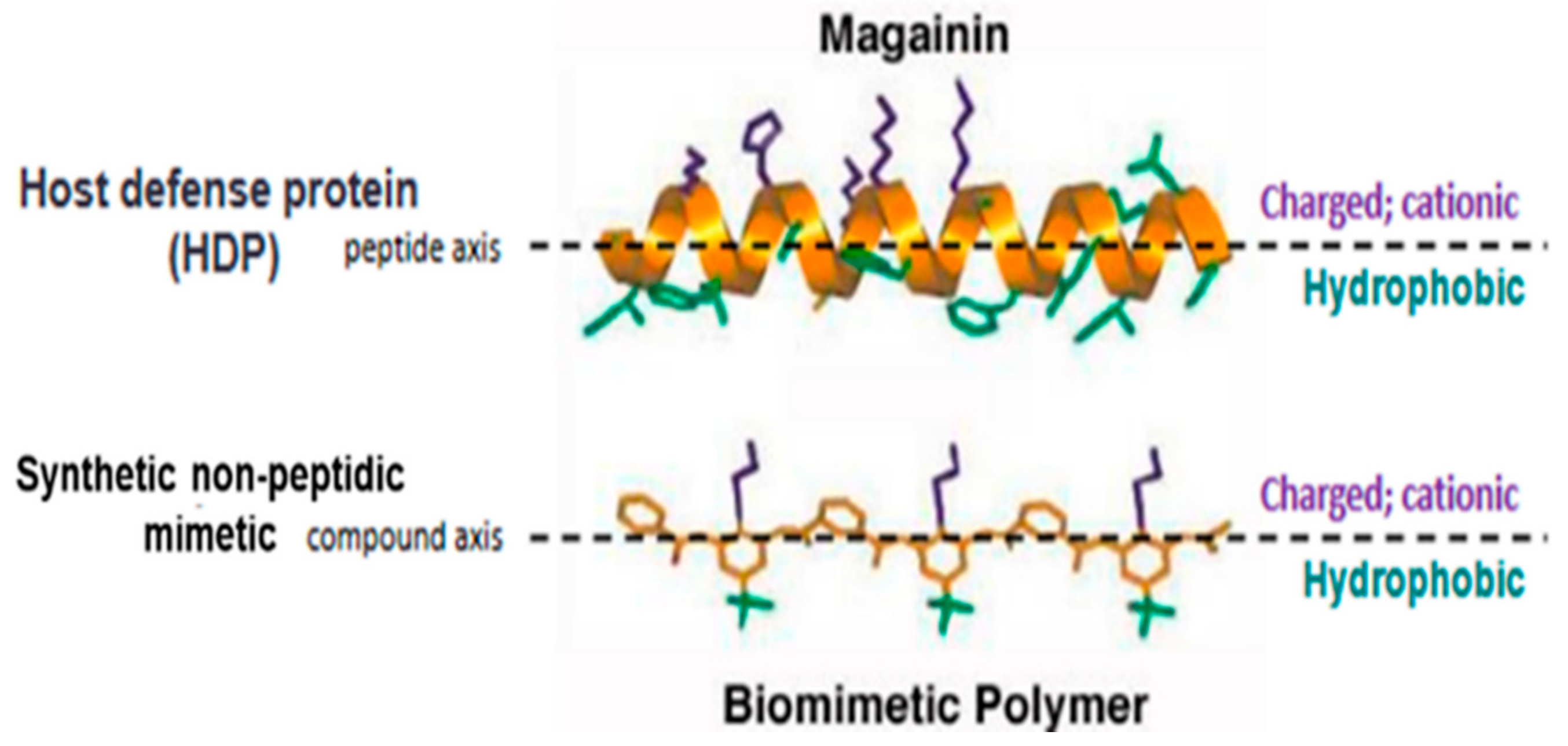{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Inhibitors
2.3. Toxicity Screens
2.4. SARS-CoV-2 Infections
2.5. Plaque Assay
2.6. RNA Extraction and RT-PCR
2.7. Statistical Analyses
3. Results
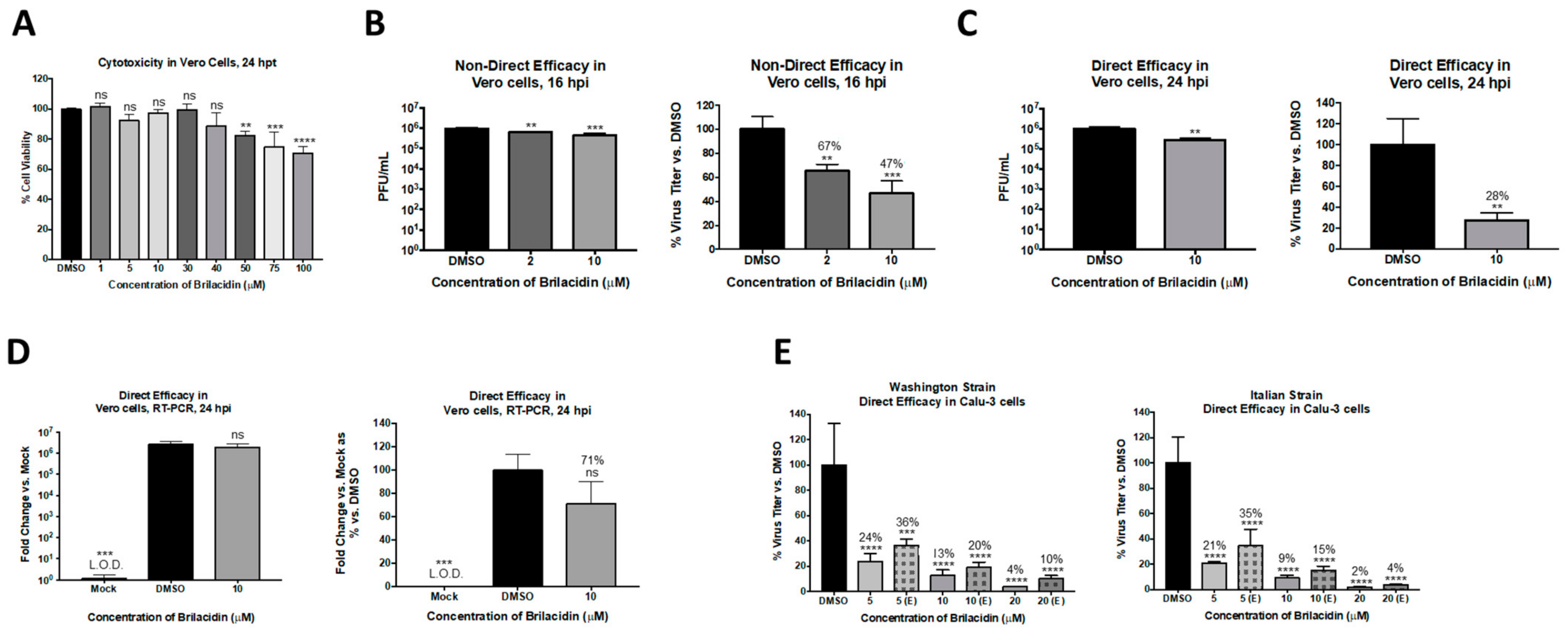
3.1. Brilacidin Inhibits SARS-CoV-2 Replication in Vero Cells
3.2. Brilacidin Appears to Disrupt the Integrity of the SARS-CoV-2 Virion in a Manner That Interferes with Entry
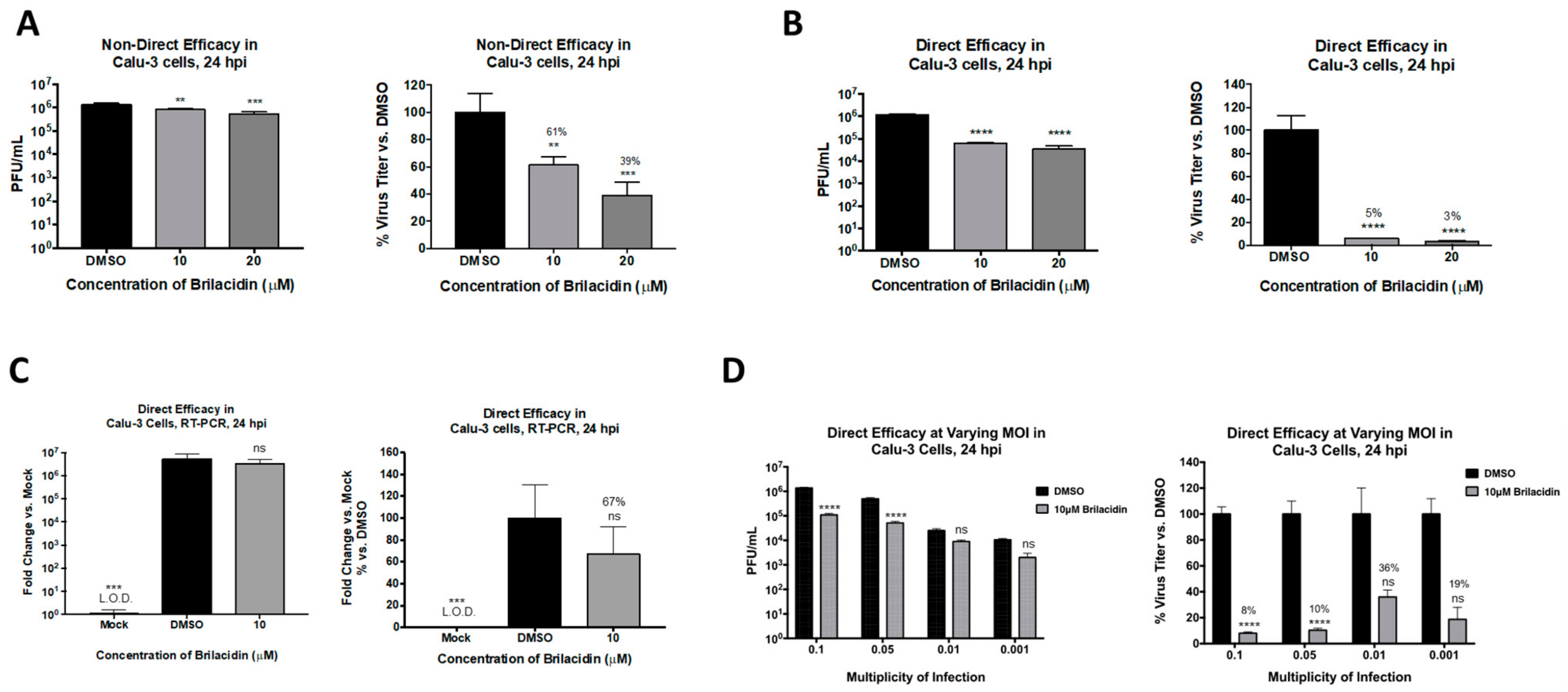
3.3. Brilacidin Inhibits SARS-CoV-2 in Calu-3 Cells
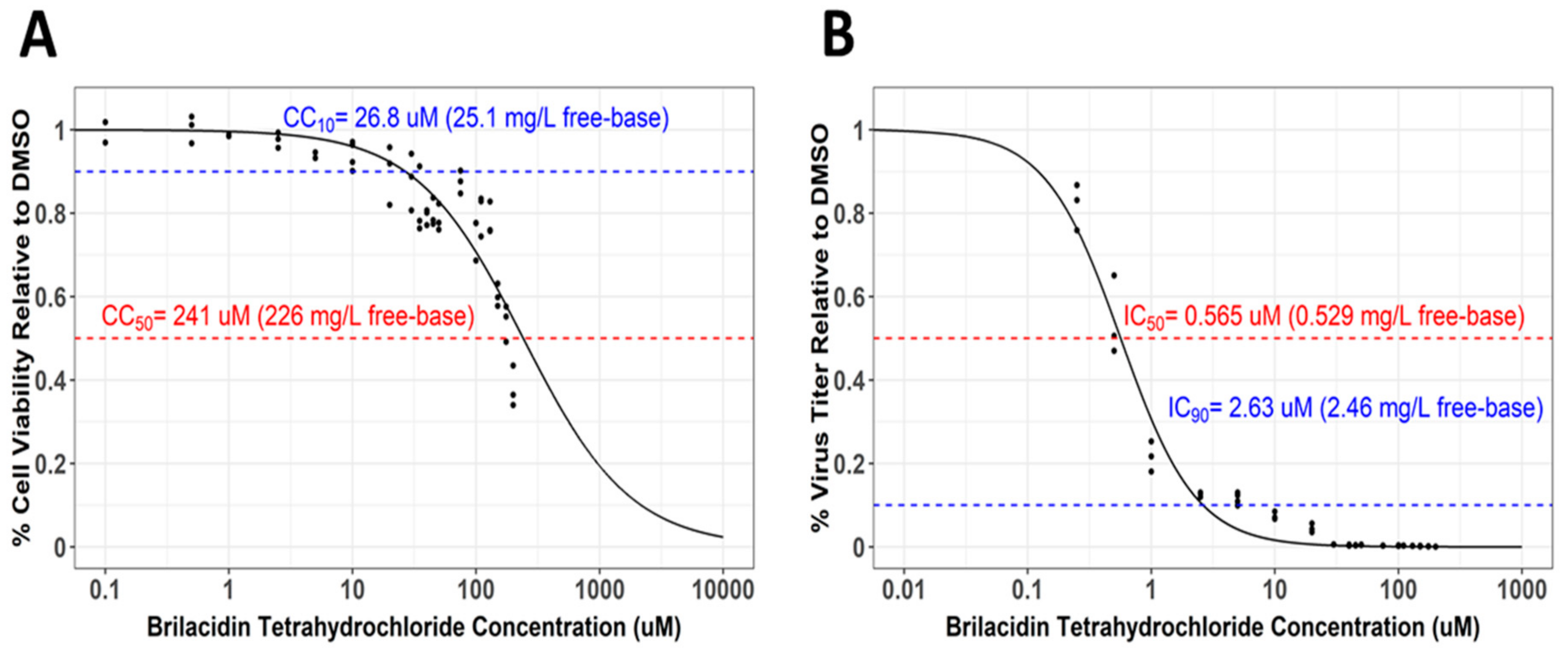
3.4. Selectivity Index Determination for Brilacidin against SARS-CoV-2 in Calu-3 Cells
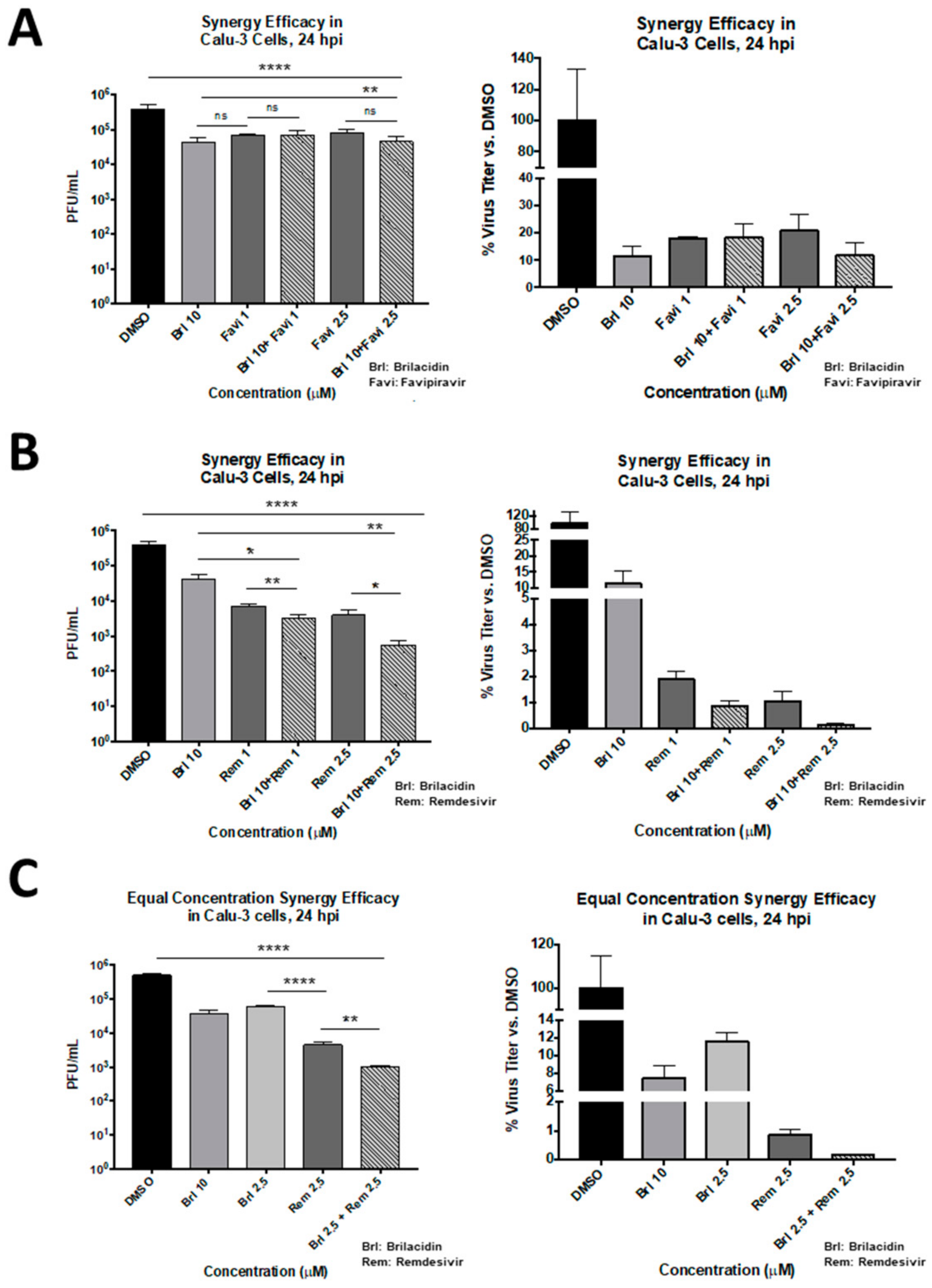
3.5. Brilacidin in Combination with Other Antiviral Treatments: Synergistic Activity against SARS-CoV-2 in Combination with Remdesivir in Calu-3 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johns Hopkins Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 27 January 2021).
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Kumar, N.; Mishra, B.; Mehmood, A.; Mohammad, A.; Mukhtar, M.S. Integrative Network Biology Framework Elucidates Molecular Mechanisms of SARS-CoV-2 Pathogenesis. iScience 2020, 23, 101526. [Google Scholar] [CrossRef]
- Hsu, A.C.-Y.; Wang, G.; Reid, A.T.; Veerati, P.C.; Pathinayake, P.S.; Daly, K.; Mayall, J.R.; Hansbro, P.M.; Horvat, J.C.; Wang, F.; et al. SARS-CoV-2 Spike protein promotes hyper-inflammatory response that can be ameliorated by Spike-antagonistic peptide and FDA-approved ER stress and MAP kinase inhibitors in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Manna, S.; Baindara, P.; Mandal, S.M. Molecular pathogenesis of secondary bacterial infection associated to viral infections including SARS-CoV-2. J. Infect. Public Health 2020, 13, 1397–1404. [Google Scholar] [CrossRef]
- Michael, J.; Cox, N.L.; Bogaert, D.; O’Grady, J. Co-infections: Potentially lethal and unexplored in COVID-19. Lancet Microbe 2020, 1, e11. [Google Scholar] [CrossRef]
- Kim, D.; Quinn, J.; Pinsky, B.; Shah, N.H.; Brown, I. Rates of Co-infection Between SARS-CoV-2 and Other Respiratory Pathogens. JAMA 2020, 323, 2085–2086. [Google Scholar] [CrossRef]
- Mirzaei, R.; Goodarzi, P.; Asadi, M.; Soltani, A.; Aljanabi, H.A.A.; Jeda, A.S.; Dashtbin, S.; Jalalifar, S.; Mohammadzadeh, R.; Teimoori, A.; et al. Bacterial co-infections with SARS-CoV-2. IUBMB Life 2020, 72, 2097–2111. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Coronavirus Disease 2019 (COVID-19). Available online: https://www.fda.gov/emergency-preparedness-and-response/counterterrorism-and-emerging-threats/coronavirus-disease-2019-covid-19 (accessed on 27 January 2021).
- Ahamad, S.; Branch, S.; Harrelson, S.; Hussain, M.K.; Saquib, M.; Khan, S. Primed for global coronavirus pandemic: Emerging research and clinical outcome. Eur. J. Med. Chem. 2020, 209, 112862. [Google Scholar] [CrossRef]
- Lythgoe, M.P.; Middleton, P. Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends Pharmacol. Sci. 2020, 41, 363–382. [Google Scholar] [CrossRef]
- Fragkou, P.C.; Belhadi, D.; Peiffer-Smadja, N.; Moschopoulos, C.D.; Lescure, F.X.; Janocha, H.; Karofylakis, E.; Yazdanpanah, Y.; Mentre, F.; Skevaki, C.; et al. Review of trials currently testing treatment and prevention of COVID-19. Clin. Microbiol. Infect. 2020, 26, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Celum, C.; Barnabas, R.; Cohen, M.S.; Collier, A.; El-Sadr, W.; Holmes, K.K.; Johnston, C.; Piot, P. Covid-19, Ebola, and HIV-Leveraging Lessons to Maximize Impact. N. Engl. J. Med. 2020, 383, e106. [Google Scholar] [CrossRef]
- Tse, L.V.; Meganck, R.M.; Graham, R.L.; Baric, R.S. The Current and Future State of Vaccines, Antivirals and Gene Therapies Against Emerging Coronaviruses. Front. Microbiol. 2020, 11, 658. [Google Scholar] [CrossRef]
- Shaman, J.; Galanti, M. Will SARS-CoV-2 become endemic? Science 2020, 370, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.I.; Ianevski, A.; Lysvand, H.; Vitkauskiene, A.; Oksenych, V.; Bjoras, M.; Telling, K.; Lutsar, I.; Dumpis, U.; Irie, Y.; et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int. J. Infect. Dis. 2020, 93, 268–276. [Google Scholar] [CrossRef]
- Al-Azzam, S.; Ding, Y.; Liu, J.; Pandya, P.; Ting, J.P.; Afshar, S. Peptides to combat viral infectious diseases. Peptides 2020, 134, 170402. [Google Scholar] [CrossRef]
- Brice, D.C.; Diamond, G. Antiviral Activities of Human Host Defense Peptides. Curr. Med. Chem. 2020, 27, 1420–1443. [Google Scholar] [CrossRef]
- Di, Y.P. Antimicrobial Peptides in Host Defense against Drug-Resistant Bacterial and Viral Infections. Curr. Med. Chem. 2020, 27, 1385–1386. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef]
- Shartouny, J.R.; Jacob, J. Mining the tree of life: Host defense peptides as antiviral therapeutics. Semin. Cell Dev. Biol. 2019, 88, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Sousa, F.H.; Casanova, V.; Stevens, C.; Barlow, P.G. Antiviral Host Defence Peptides. In Host Defense Peptides and Their Potential as Therapeutic Agents; Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chou, Y.Y.; Chang, T.L. Defensins in viral infections. J. Innate Immun. 2009, 1, 413–420. [Google Scholar] [CrossRef]
- Klotman, M.E.; Chang, T.L. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Belanger, C.R.; Haney, E.F.; Hancock, R.E.W. 10-Host defense (antimicrobial) peptides. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Sawston, UK, 2018; pp. 253–285. [Google Scholar] [CrossRef]
- Scott, R.W.; Tew, G.N. Mimics of Host Defense Proteins; Strategies for Translation to Therapeutic Applications. Curr. Top. Med. Chem. 2017, 17, 576–589. [Google Scholar] [CrossRef]
- Research Topic: Advances in the Immunology of Host Defense Peptide: Mechanisms and Applications of Antimicrobial Functions and Beyond. (2019–2020). Front. Immunol. 2020. Available online: https://www.frontiersin.org/research-topics/11137/advances-in-the-immunology-of-host-defense-peptide-mechanisms-and-applications-of-antimicrobial-func (accessed on 5 February 2021).
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. The antimicrobial peptide database provides a platform for decoding the design principles of naturally occurring antimicrobial peptides. Protein Sci. 2020, 29, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides With Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef]
- Brender, J.R.; McHenry, A.J.; Ramamoorthy, A. Does cholesterol play a role in the bacterial selectivity of antimicrobial peptides? Front. Immunol. 2012, 3, 195. [Google Scholar] [CrossRef] [PubMed]
- Goluszko, P.; Nowicki, B. Membrane cholesterol: A crucial molecule affecting interactions of microbial pathogens with mammalian cells. Infect. Immun. 2005, 73, 7791–7796. [Google Scholar] [CrossRef]
- Salata, C.; Calistri, A.; Parolin, C.; Baritussio, A.; Palu, G. Antiviral activity of cationic amphiphilic drugs. Expert Rev. Anti-infect. Ther. 2017, 15, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Idris, M.M.; Banu, S.; Siva, A.B.; Nagaraj, R. Downregulation of Defensin genes in SARS-CoV-2 infection. medRxiv 2020. [Google Scholar] [CrossRef]
- Pahar, B.; Madonna, S.; Das, A.; Albanesi, C.; Girolomoni, G. Immunomodulatory Role of the Antimicrobial LL-37 Peptide in Autoimmune Diseases and Viral Infections. Vaccines 2020, 8, 517. [Google Scholar] [CrossRef] [PubMed]
- Crane-Godreau, M.A.; Clem, K.J.; Payne, P.; Fiering, S. Vitamin D Deficiency and Air Pollution Exacerbate COVID-19 Through Suppression of Antiviral Peptide LL37. Front. Public Health 2020, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef]
- Mahendran, A.S.K.; Lim, Y.S.; Fang, C.M.; Loh, H.S.; Le, C.F. The Potential of Antiviral Peptides as COVID-19 Therapeutics. Front. Pharmacol. 2020, 11, 575444. [Google Scholar] [CrossRef]
- VanPatten, S.; He, M.; Altiti, A.; Cheng, K.F.; Ghanem, M.H.; Al-Abed, Y. Evidence supporting the use of peptides and peptidomimetics as potential SARS-CoV-2 (COVID-19) therapeutics. Future Med. Chem. 2020, 12, 1647–1656. [Google Scholar] [CrossRef]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.N.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Memariani, H.; Memariani, M. Therapeutic and prophylactic potential of anti-microbial peptides against coronaviruses. Ir. J. Med. Sci. 2020, 189, 1153–1154. [Google Scholar] [CrossRef]
- Whisenant, J.; Burgess, K. Blocking Coronavirus 19 Infection via the SARS-CoV-2 Spike Protein: Initial Steps. ACS Med. Chem. Lett. 2020, 11, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Li, D.; Zhao, X.; Han, S.; Wang, T.; Zhao, G.; Chen, Y.; Chen, F.; Zhao, J.; et al. Lectin-like Intestinal Defensin Inhibits 2019-nCoV Spike binding to ACE2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Barh, D.T.; Tiwari, S.; Andrade, B.; Giovanetti, M.; Kumavath, R.; Ghosh, P.; Góes-Neto, A.; Carlos, A.L., Jr.; Azevedo, V. Potential Chimeric Peptides to Block the SARS-CoV-2 Spike RBD. Preprints 2020, 1. [Google Scholar] [CrossRef]
- Panda, S.K.G.; Gupta, P.S.S.; Biswal, S.; Ray, A.K.; Rana, M.K. ACE-2-derived Biomimetic Peptides for the Inhibition of Spike Protein of SARS-CoV-2. ChemRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Chen, B.; Tian, E.K.; He, B.; Tian, L.; Han, R.; Wang, S.; Xiang, Q.; Zhang, S.; El Arnaout, T.; Cheng, W. Overview of lethal human coronaviruses. Signal Transduct. Target. Ther. 2020, 5, 89. [Google Scholar] [CrossRef]
- Som, A.; Navasa, N.; Percher, A.; Scott, R.W.; Tew, G.N.; Anguita, J. Identification of synthetic host defense peptide mimics that exert dual antimicrobial and anti-inflammatory activities. Clin. Vaccine Immunol. 2012, 19, 1784–1791. [Google Scholar] [CrossRef]
- Tew, G.N.; Scott, R.W.; Klein, M.L.; Degrado, W.F. De novo design of antimicrobial polymers, foldamers, and small molecules: From discovery to practical applications. Acc. Chem. Res. 2010, 43, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar] [CrossRef]
- Scott, R.W.; DeGrado, W.F.; Tew, G.N. De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotechnol. 2008, 19, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Tew, G.N.; Liu, D.; Chen, B.; Doerksen, R.J.; Kaplan, J.; Carroll, P.J.; Klein, M.L.; DeGrado, W.F. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 5110–5114. [Google Scholar] [CrossRef] [PubMed]
- New Weapons for the Germ Wars: Inexpensive Polymers Can Extend the Range of Nature’s Germ-Fighter Arsenal. In Projects in Scientific Computing; University of Pittsburgh Supercomputing Center: Pittsburgh, PA, USA, 2002; Available online: http://www.ipharminc.com/s/new_weapons_for_the_germ_wars.pdf (accessed on 27 January 2021).
- Langreth, R. Antibiotic Artisan. Forbes, 26 January 2011. [Google Scholar]
- Ergene, C.; Yasuhara, K.; Palermo, E.F. Biomimetic antimicrobial polymers: Recent advances in molecular design. Polym. Chem. 2018, 9, 2407–2427. [Google Scholar] [CrossRef]
- Gopalakrishnan, R.; Frolov, A.I.; Knerr, L.; Drury, W.J.; Valeur, E. Therapeutic Potential of Foldamers: From Chemical Biology Tools To Drug Candidates? J. Med. Chem. 2016, 59, 9599–9621. [Google Scholar] [CrossRef]
- Nizami, B.; Bereczki-Szakál, D.; Varró, N.; El Battioui, K.; Nagaraj, V.U.; Szigyártó, I.C.; Mándity, I.; Beke-Somfai, T. FoldamerDB: A database of peptidic foldamers. Nucleic Acids Res. 2019, 48, D1122–D1128. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef]
- Brilacidin as a Successful Example of de Novo Drug Design; Innovation Pharmaceuticals: Wakefield, MA, USA, 2017.
- Korendovych, I.V.; DeGrado, W.F. De novo protein design, a retrospective. Q. Rev. Biophys. 2020, 53, e3. [Google Scholar] [CrossRef] [PubMed]
- Hutson, M. Annals of Technology: Scientists Advance on One of Technology’s Holy Grails: Designer Proteins Could Help Us Build New Materials, Clean up the Environment, and Even Fight COVID-19. The New Yorker, 18 September 2020. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Claudio, C.; Juan Di, F. In silico Drug Repurposing for COVID-19: Targeting SARS-CoV-2 Proteins through Docking and Quantum Mechanical Scoring; American Chemical Society: Washington, DC, USA, 2020. [Google Scholar] [CrossRef]
- COVID-19 Worldwide Preclinical Studies: Preclinical In Vitro Studies. Available online: https://ghddi-ailab.github.io/Targeting2019-nCoV/preclinical/ (accessed on 17 October 2020).
- Ellinger, B.; Bojkova, D.; Zaliani, A.; Cinatl, J.; Claussen, C.; Westhaus, S.; Reinshagen, J.; Kuzikov, M.; Wolf, M.; Geisslinger, G.; et al. Identification of inhibitors of SARS-CoV-2 in-vitro cellular toxicity in human (Caco-2) cells using a large scale drug repurposing collection. Preprints 2020. [Google Scholar] [CrossRef]
- Cao, B.; Hayden, F.G. Antiviral monotherapy for hospitalised patients with COVID-19 is not enough. Lancet 2020, 396, 1310–1311. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Kar, S.; Aliter, K.F. Potential Anti-COVID-19 Therapeutics that Block the Early Stage of the Viral Life Cycle: Structures, Mechanisms, and Clinical Trials. Int. J. Mol. Sci. 2020, 21, 5224. [Google Scholar] [CrossRef] [PubMed]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar] [CrossRef]
- Su, X.; Wang, Q.; Wen, Y.; Jiang, S.; Lu, L. Protein- and Peptide-Based Virus Inactivators: Inactivating Viruses Before Their Entry Into Cells. Front. Microbiol. 2020, 11, 1063. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakovic, A.; Risner, K.; Bhalla, N.; Alem, F.; Chang, T.L.; Weston, W.K.; Harness, J.A.; Narayanan, A. Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture. Viruses 2021, 13, 271. https://doi.org/10.3390/v13020271
Bakovic A, Risner K, Bhalla N, Alem F, Chang TL, Weston WK, Harness JA, Narayanan A. Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture. Viruses. 2021; 13(2):271. https://doi.org/10.3390/v13020271
Chicago/Turabian StyleBakovic, Allison, Kenneth Risner, Nishank Bhalla, Farhang Alem, Theresa L. Chang, Warren K. Weston, Jane A. Harness, and Aarthi Narayanan. 2021. "Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture" Viruses 13, no. 2: 271. https://doi.org/10.3390/v13020271
APA StyleBakovic, A., Risner, K., Bhalla, N., Alem, F., Chang, T. L., Weston, W. K., Harness, J. A., & Narayanan, A. (2021). Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture. Viruses, 13(2), 271. https://doi.org/10.3390/v13020271