
Advances in Hepatitis E Virus Biology and Pathogenesis


Abstract
1. Introduction
2. HEV Taxonomy and Distribution
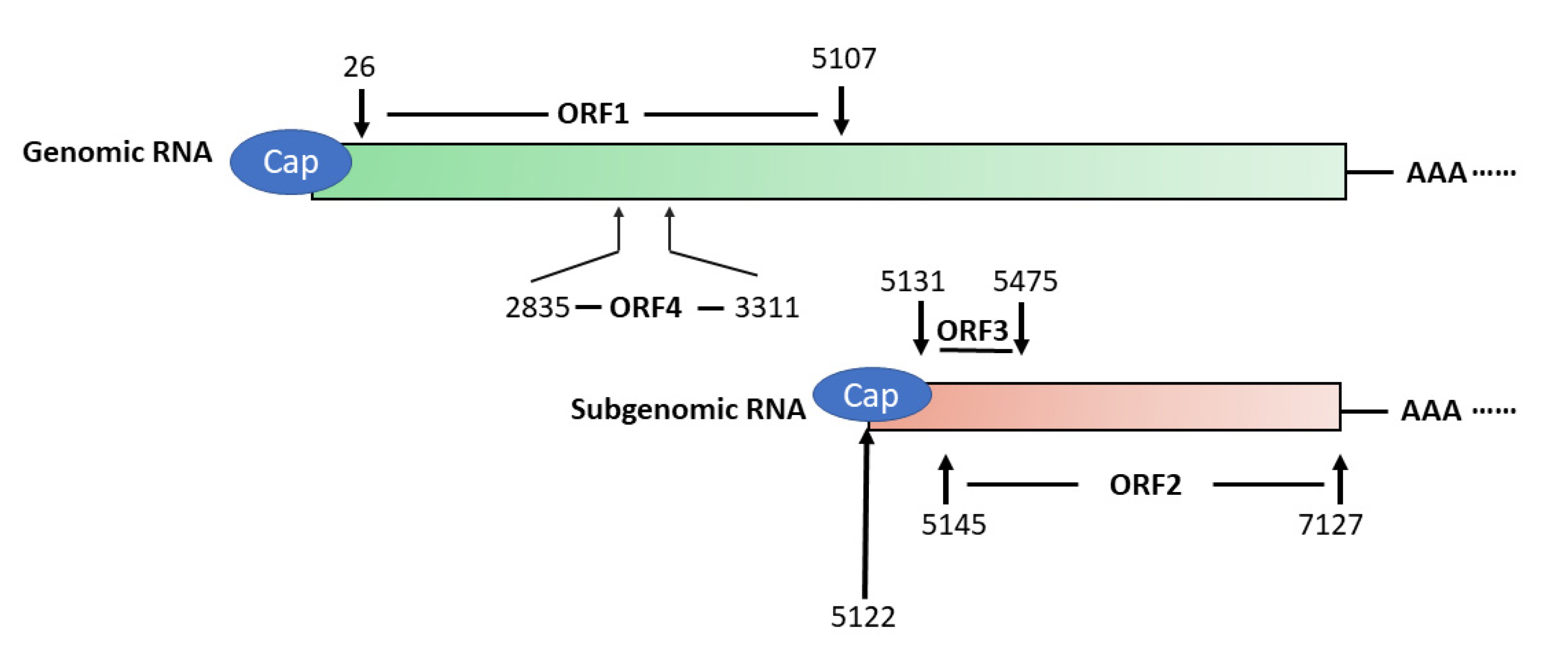
3. HEV Genome and its Encoded Proteins
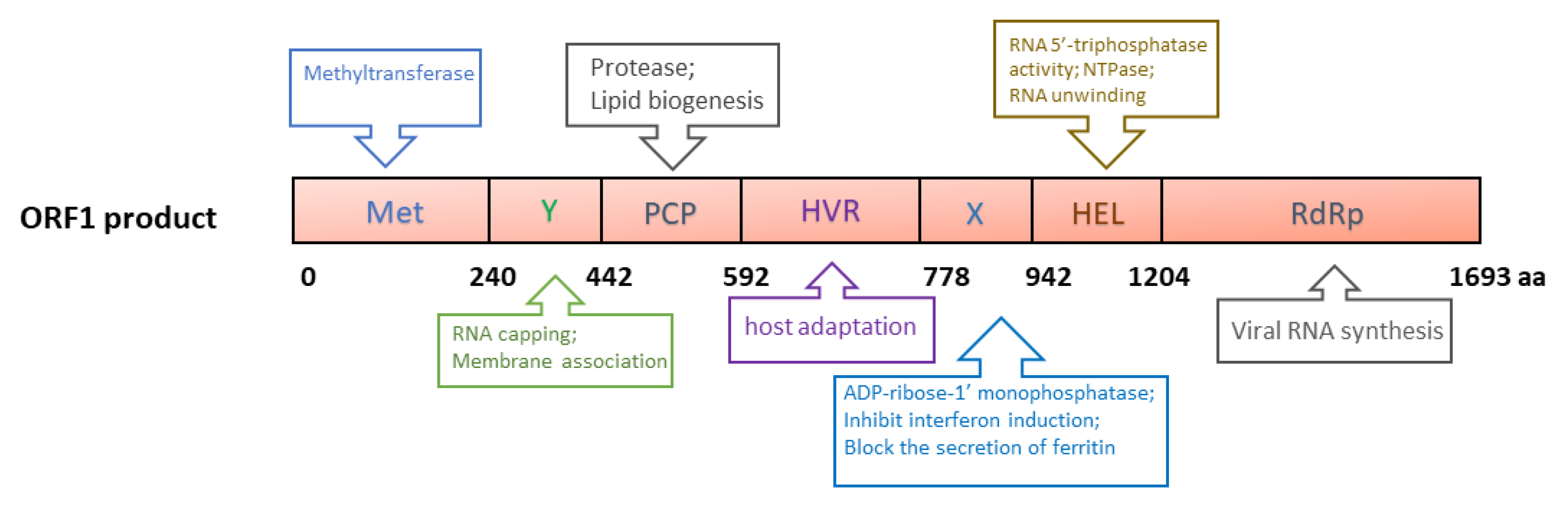
3.1. ORF1
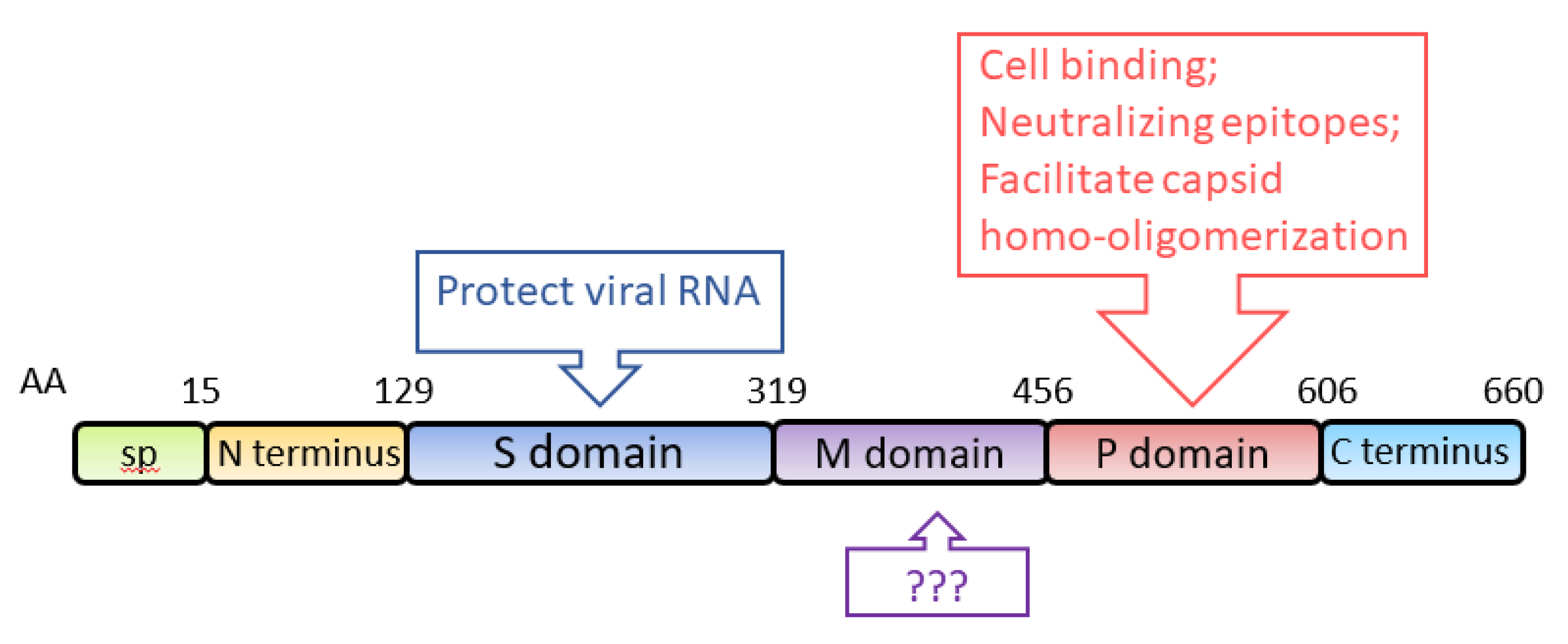
3.2. ORF2
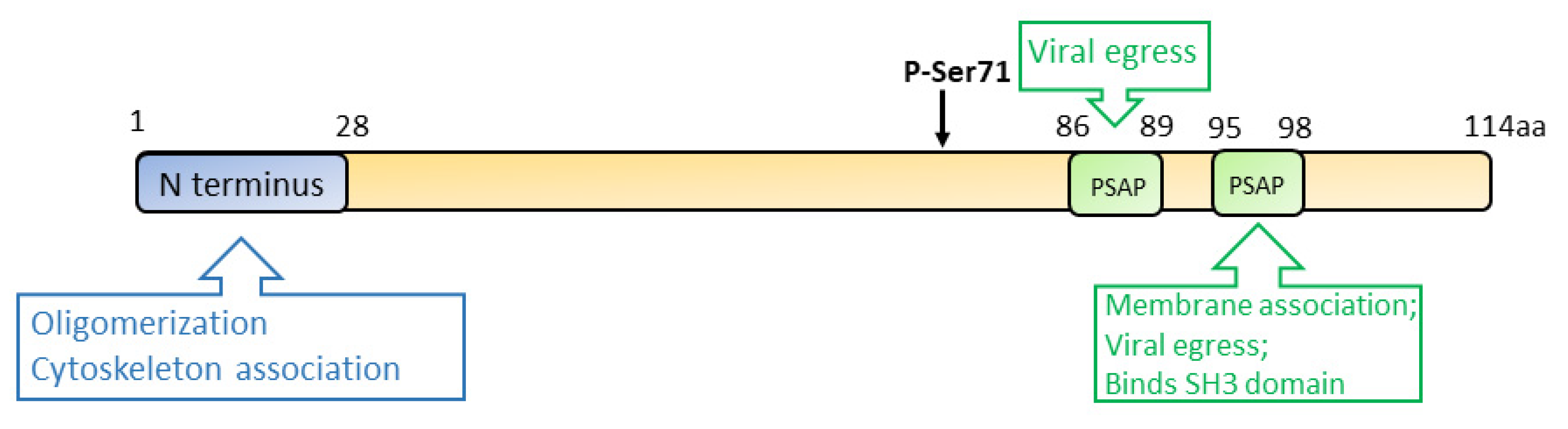
3.3. ORF3
3.4. ORF4
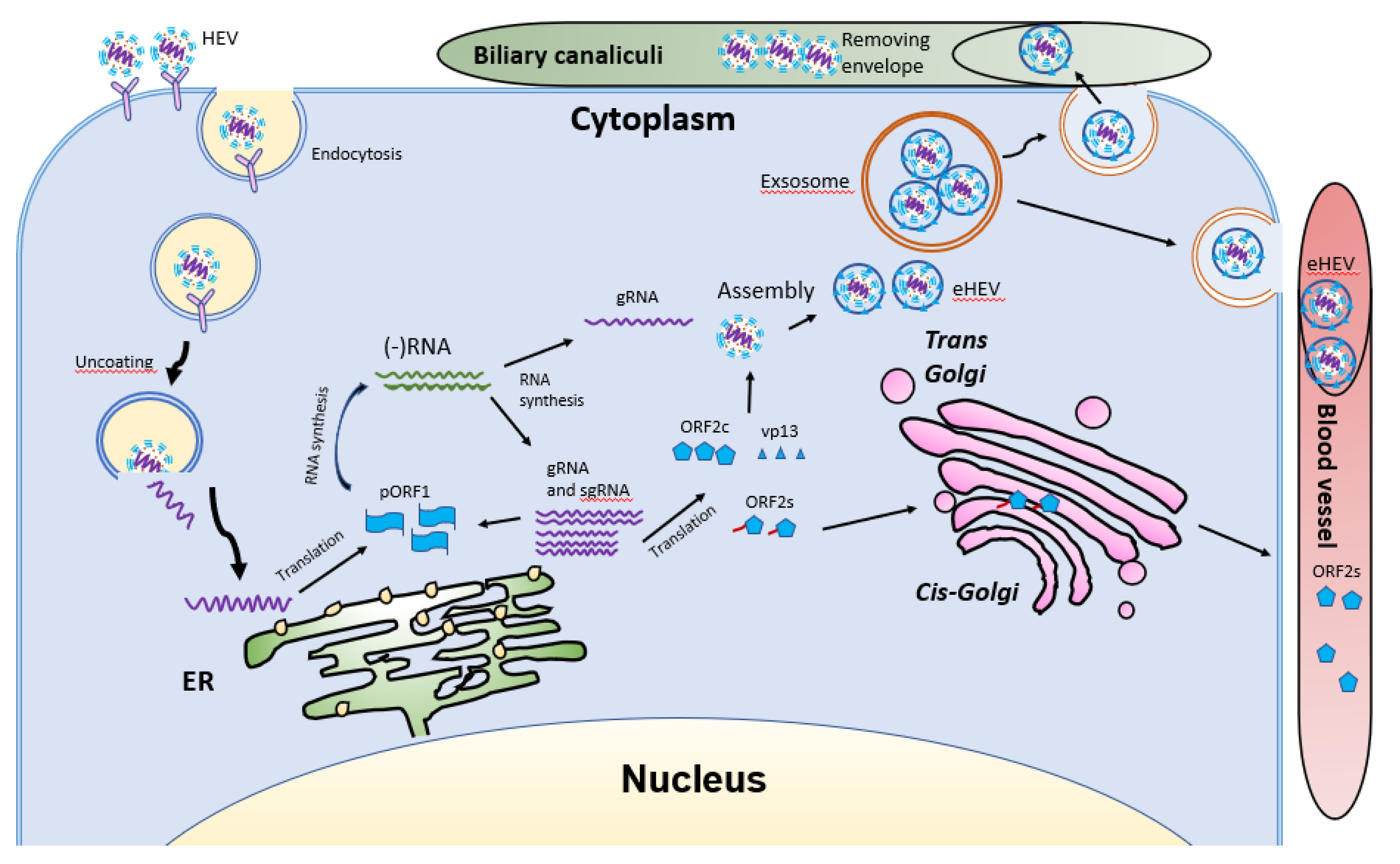
4. HEV Life Cycle
5. Manipulation of Host Factors by HEV for Its Replication
5.1. Interference with Innate Immune Response
5.1.1. Interplay with Type I IFNs
5.1.2. Induction of Type II IFN
5.1.3. Interplay with Type III IFNs
5.2. Manipulation of Cellular Factors for a Conducive Environment for Replication
6. Clinical Manifestations of Hepatitis E
7. HEV Infection of Pregnant Women
8. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO; Hepatitis, E. Available online: https://www.who.int/immunization/diseases/hepatitisE/en/ (accessed on 20 December 2020).
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Purdy, M.A.; International Committee on the Taxonomy of Viruses Hepeviridae Study Group. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Jirintai; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E Virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: Characterization of HEV virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Tanggis; Kobayashi, T.; Nishizawa, T.; Okamoto, H. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-Golgi network protein 2. Arch. Virol. 2014, 159, 979–991. [Google Scholar] [CrossRef]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Smith, D.B.; Ictv Report, C. ICTV Virus Taxonomy Profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J. Recent advances in Hepatitis E virus. J. Viral Hepat. 2010, 17, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kantala, T.; Heinonen, M.; Oristo, S.; von Bonsdorff, C.H.; Maunula, L. Hepatitis E virus in young pigs in Finland and characterization of the isolated partial genomic sequences of genotype 3 HEV. Foodborne Pathog. Dis. 2015, 12, 253–260. [Google Scholar] [CrossRef]
- Merino-Ramos, T.; Martin-Acebes, M.A.; Casal, J.; Saiz, J.C.; Loza-Rubio, E. Prevalence of Hepatitis E Virus (HEV) Antibodies in Mexican Pigs. Food Environ. Virol. 2016, 8, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Modesto, P.; Bertolini, S.; Peletto, S.; Acutis, P.L.; Dondo, A.; Robetto, S.; Mignone, W.; Orusa, R.; Ru, G.; et al. Serological and virological survey of hepatitis E virus in wild boar populations in northwestern Italy: Detection of HEV subtypes 3e and 3f. Arch. Virol. 2015, 160, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Dubois, M.; Abravanel, F.; Top, S.; Bertagnoli, S.; Guerin, J.L.; Izopet, J. Risk of zoonotic transmission of HEV from rabbits. J. Clin. Virol. 2013, 58, 357–362. [Google Scholar] [CrossRef]
- Huang, F.; Li, Y.; Yu, W.; Jing, S.; Wang, J.; Long, F.; He, Z.; Yang, C.; Bi, Y.; Cao, W.; et al. Excretion of infectious hepatitis E virus into milk in cows imposes high risks of zoonosis. Hepatology 2016, 64, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Nidaira, M.; Takahashi, K.; Ogura, G.; Taira, K.; Okano, S.; Kudaka, J.; Itokazu, K.; Mishiro, S.; Nakamura, M. Detection and phylogenetic analysis of hepatitis E viruses from mongooses in Okinawa, Japan. J. Vet. Med. Sci. 2012, 74, 1665–1668. [Google Scholar] [CrossRef]
- Sarchese, V.; Di Profio, F.; Melegari, I.; Palombieri, A.; Sanchez, S.B.; Arbuatti, A.; Ciuffetelli, M.; Marsilio, F.; Martella, V.; Di Martino, B. Hepatitis E virus in sheep in Italy. Transbound. Emerg. Dis. 2019, 66, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Takeda, N.; Muramatsu, M.; Li, T.C. High Prevalence of Hepatitis E Virus Infection in Imported Cynomolgus Monkeys in Japan. Jpn. J. Infect. Dis. 2019, 72, 429–431. [Google Scholar] [CrossRef]
- Anheyer-Behmenburg, H.E.; Szabo, K.; Schotte, U.; Binder, A.; Klein, G.; Johne, R. Hepatitis E Virus in Wild Boars and Spillover Infection in Red and Roe Deer, Germany, 2013-2015. Emerg. Infect. Dis. 2017, 23, 130–133. [Google Scholar] [CrossRef]
- Takahashi, K.; Kitajima, N.; Abe, N.; Mishiro, S. Complete or near-complete nucleotide sequences of hepatitis E virus genome recovered from a wild boar, a deer, and four patients who ate the deer. Virology 2004, 330, 501–505. [Google Scholar] [CrossRef]
- Di Pasquale, S.; De Santis, P.; La Rosa, G.; Di Domenico, K.; Iaconelli, M.; Micarelli, G.; Martini, E.; Bilei, S.; De Medici, D.; Suffredini, E. Quantification and genetic diversity of Hepatitis E virus in wild boar (Sus scrofa) hunted for domestic consumption in Central Italy. Food Microbiol. 2019, 82, 194–201. [Google Scholar] [CrossRef]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357. [Google Scholar] [CrossRef]
- Sridhar, S.; Teng, J.L.L.; Chiu, T.H.; Lau, S.K.P.; Woo, P.C.Y. Hepatitis E Virus Genotypes and Evolution: Emergence of Camel Hepatitis E Variants. Int. J. Mol. Sci. 2017, 18, 869. [Google Scholar] [CrossRef]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.; Cai, J.; Zhang, A.J.; Leung, K.H.; Chung, T.W.H.; Chan, J.F.W.; Chan, W.M.; Teng, J.L.L.; et al. Rat Hepatitis E Virus as Cause of Persistent Hepatitis after Liver Transplant. Emerg. Infect. Dis. 2018, 24, 2241–2250. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, A.; Pankovics, P. Review of Hepatitis E Virus in Rats: Evident Risk of Species Orthohepevirus C to Human Zoonotic Infection and Disease. Viruses 2020, 12, 1148. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Harms, D.; Yang, X.L.; Bock, C.T. Orthohepevirus C: An Expanding Species of Emerging Hepatitis E Virus Variants. Pathogens 2020, 9, 154. [Google Scholar] [CrossRef]
- Nelson, K.E.; Labrique, A.B.; Kmush, B.L. Epidemiology of Genotype 1 and 2 Hepatitis E Virus Infections. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Akanbi, O.A.; Harms, D.; Wang, B.; Opaleye, O.O.; Adesina, O.; Osundare, F.A.; Ogunniyi, A.; Naidoo, D.; Devaux, I.; Wondimagegnehu, A.; et al. Complete Genome Sequence of a Hepatitis E Virus Genotype 1e Strain from an Outbreak in Nigeria, 2017. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Nguyen, D.; Fernandez, J.; Yun, K.Y.; Fry, K.E.; Bradley, D.W.; Tam, A.W.; Reyes, G.R. Molecular cloning and sequencing of the Mexico isolate of hepatitis E virus (HEV). Virology 1992, 191, 550–558. [Google Scholar] [CrossRef]
- Bouscaillou, J.; Komas, N.; Tricou, V.; Nakoune, E.; Selekon, B.; Fontanet, A.; Kazanji, M. Imported hepatitis e virus, central african republic, 2011. Emerg. Infect. Dis. 2013, 19, 335–337. [Google Scholar] [CrossRef]
- Kaiser, M.; Kamili, S.; Hayden, T.; Blumel, J.; Baylis, S.A. Genome Sequence of a Genotype 2 Hepatitis E Virus World Health Organization Reference Strain. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Wang, B.; Akanbi, O.A.; Harms, D.; Adesina, O.; Osundare, F.A.; Naidoo, D.; Deveaux, I.; Ogundiran, O.; Ugochukwu, U.; Mba, N.; et al. A new hepatitis E virus genotype 2 strain identified from an outbreak in Nigeria, 2017. Virol. J. 2018, 15, 163. [Google Scholar] [CrossRef]
- Kamar, N.; Pischke, S. Acute and Persistent Hepatitis E Virus Genotype 3 and 4 Infection: Clinical Features, Pathogenesis, and Treatment. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Li, T.C.; Ochiai, S.; Ishiko, H.; Wakita, T.; Miyamura, T.; Takeda, N. A retrospective study on imported hepatitis E in Japan. Travel Med. Infect. Dis. 2012, 10, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Adlhoch, C.; Avellon, A.; Baylis, S.A.; Ciccaglione, A.R.; Couturier, E.; de Sousa, R.; Epstein, J.; Ethelberg, S.; Faber, M.; Feher, A.; et al. Hepatitis E virus: Assessment of the epidemiological situation in humans in Europe, 2014/15. J. Clin. Virol. 2016, 82, 9–16. [Google Scholar] [CrossRef] [PubMed]
- de la Caridad Montalvo Villalba, M.; Owot, J.C.; Benedito, E.C.; Corredor, M.B.; Flaquet, P.P.; Frometa, S.S.; Wong, M.S.; Rodriguez Lay Lde, L. Hepatitis E virus genotype 3 in humans and swine, Cuba. Infect. Genet. Evol. 2013, 14, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Fierro, N.A.; Realpe, M.; Meraz-Medina, T.; Roman, S.; Panduro, A. Hepatitis E virus: An ancient hidden enemy in Latin America. World J. Gastroenterol. 2016, 22, 2271–2283. [Google Scholar] [CrossRef]
- Si, F.; Shi, B.; Wang, X.; Zhu, Y.; Liu, X.; Yang, Q.; Li, Z. Construction of an infectious cDNA clone of a swine genotype 3 HEV strain isolated in Shanghai, China. Intervirology 2014, 57, 74–82. [Google Scholar] [CrossRef]
- Pavio, N.; Meng, X.J.; Doceul, V. Zoonotic origin of hepatitis E. Curr. Opin. Virol. 2015, 10, 34–41. [Google Scholar] [CrossRef]
- Colson, P.; Borentain, P.; Queyriaux, B.; Kaba, M.; Moal, V.; Gallian, P.; Heyries, L.; Raoult, D.; Gerolami, R. Pig liver sausage as a source of hepatitis E virus transmission to humans. J. Infect. Dis. 2010, 202, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Park, B.J.; Kim, Y.H.; Choi, Y.S.; Ahn, H.S.; Han, S.H.; Choi, I.S. Isolation of hepatitis E virus genotype 4 from patients with acute cryptogenic hepatitis in Korea. J. Clin. Virol. 2017, 89, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Tesse, S.; Lioure, B.; Fornecker, L.; Wendling, M.J.; Stoll-Keller, F.; Bigaillon, C.; Nicand, E. Circulation of genotype 4 hepatitis E virus in Europe: First autochthonous hepatitis E infection in France. J. Clin. Virol. 2012, 54, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Yue, N.; Wang, Q.; Zheng, M.; Wang, D.; Duan, C.; Yu, X.; Zhang, X.; Bao, C.; Jin, H. Prevalence of hepatitis E virus infection among people and swine in mainland China: A systematic review and meta-analysis. Zoonoses Public Health 2019, 66, 265–275. [Google Scholar] [CrossRef]
- Takahashi, M.; Nishizawa, T.; Yoshikawa, A.; Sato, S.; Isoda, N.; Ido, K.; Sugano, K.; Okamoto, H. Identification of two distinct genotypes of hepatitis E virus in a Japanese patient with acute hepatitis who had not travelled abroad. J. Gen. Virol. 2002, 83, 1931–1940. [Google Scholar] [CrossRef]
- Ticehurst, J.R.; Pisanic, N.; Forman, M.S.; Ordak, C.; Heaney, C.D.; Ong, E.; Linnen, J.M.; Ness, P.M.; Guo, N.; Shan, H.; et al. Probable transmission of hepatitis E virus (HEV) via transfusion in the United States. Transfusion 2019, 59, 1024–1034. [Google Scholar] [CrossRef]
- Sridhar, S.; Cheng, V.C.C.; Wong, S.C.; Yip, C.C.Y.; Wu, S.; Lo, A.W.I.; Leung, K.H.; Mak, W.W.N.; Cai, J.; Li, X.; et al. Donor-Derived Genotype 4 Hepatitis E Virus Infection, Hong Kong, China, 2018. Emerg. Infect. Dis. 2019, 25, 425–433. [Google Scholar] [CrossRef]
- Tripathy, A.S.; Puranik, S.; Sharma, M.; Chakraborty, S.; Devakate, U.R. Hepatitis E virus seroprevalence among blood donors in Pune, India. J. Med. Virol. 2019, 91, 813–819. [Google Scholar] [CrossRef]
- Meng, J.; Dai, X.; Chang, J.C.; Lopareva, E.; Pillot, J.; Fields, H.A.; Khudyakov, Y.E. Identification and characterization of the neutralization epitope(s) of the hepatitis E virus. Virology 2001, 288, 203–211. [Google Scholar] [CrossRef]
- Emerson, S.U.; Clemente-Casares, P.; Moiduddin, N.; Arankalle, V.A.; Torian, U.; Purcell, R.H. Putative neutralization epitopes and broad cross-genotype neutralization of Hepatitis E virus confirmed by a quantitative cell-culture assay. J. Gen. Virol. 2006, 87, 697–704. [Google Scholar] [CrossRef]
- Engle, R.E.; Yu, C.; Emerson, S.U.; Meng, X.J.; Purcell, R.H. Hepatitis E virus (HEV) capsid antigens derived from viruses of human and swine origin are equally efficient for detecting anti-HEV by enzyme immunoassay. J. Clin. Microbiol. 2002, 40, 4576–4580. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, K.; Yamada, K.; Tanaka, T.; Nagashima, S.; Jirintai; Takahashi, M.; Okamoto, H. Determination of the 5′-terminal sequence of subgenomic RNA of hepatitis E virus strains in cultured cells. Arch. Virol. 2009, 154, 1945–1951. [Google Scholar] [CrossRef]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef]
- Nan, Y.; Zhang, Y.J. Molecular Biology and Infection of Hepatitis E Virus. Front. Microbiol. 2016, 7, 1419. [Google Scholar] [CrossRef]
- Sehgal, D.; Thomas, S.; Chakraborty, M.; Jameel, S. Expression and processing of the Hepatitis E virus ORF1 nonstructural polyprotein. Virol. J. 2006, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K. Molecular characterization of hepatitis E virus ORF1 gene supports a papain-like cysteine protease (PCP)-domain activity. Virus Res. 2013, 178, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Szkolnicka, D.; Pollan, A.; Da Silva, N.; Oechslin, N.; Gouttenoire, J.; Moradpour, D. Recombinant Hepatitis E Viruses Harboring Tags in the ORF1 Protein. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Karlin, D.G. Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol. Direct. 2015, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Magden, J.; Takeda, N.; Li, T.; Auvinen, P.; Ahola, T.; Miyamura, T.; Merits, A.; Kaariainen, L. Virus-specific mRNA capping enzyme encoded by hepatitis E virus. J. Virol. 2001, 75, 6249–6255. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Lole, K.S. NTPase and 5′ to 3′ RNA duplex-unwinding activities of the hepatitis E virus helicase domain. J. Virol. 2010, 84, 3595–3602. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K. The hepatitis E virus ORF1 ‘X-domain’ residues form a putative macrodomain protein/Appr-1″-pase catalytic-site, critical for viral RNA replication. Gene 2015, 566, 47–53. [Google Scholar] [CrossRef]
- Mahilkar, S.; Paingankar, M.S.; Lole, K.S. Hepatitis E virus RNA-dependent RNA polymerase: RNA template specificities, recruitment and synthesis. J. Gen. Virol. 2016, 97, 2231–2242. [Google Scholar] [CrossRef]
- LeDesma, R.; Nimgaonkar, I.; Ploss, A. Hepatitis E Virus Replication. Viruses 2019, 11, 719. [Google Scholar] [CrossRef]
- Li, T.C.; Yamakawa, Y.; Suzuki, K.; Tatsumi, M.; Razak, M.A.; Uchida, T.; Takeda, N.; Miyamura, T. Expression and self-assembly of empty virus-like particles of hepatitis E virus. J. Virol. 1997, 71, 7207–7213. [Google Scholar] [CrossRef]
- Li, T.C.; Takeda, N.; Miyamura, T.; Matsuura, Y.; Wang, J.C.; Engvall, H.; Hammar, L.; Xing, L.; Cheng, R.H. Essential elements of the capsid protein for self-assembly into empty virus-like particles of hepatitis E virus. J. Virol. 2005, 79, 12999–13006. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Wang, J.C.; Li, T.C.; Yasutomi, Y.; Lara, J.; Khudyakov, Y.; Schofield, D.; Emerson, S.U.; Purcell, R.H.; Takeda, N.; et al. Spatial configuration of hepatitis E virus antigenic domain. J. VIrol. 2011, 85, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Mori, Y.; Miyazaki, N.; Cheng, R.H.; Yoshimura, M.; Unno, H.; Shima, R.; Moriishi, K.; Tsukihara, T.; Li, T.C.; et al. Biological and immunological characteristics of hepatitis E virus-like particles based on the crystal structure. Proc. Natl. Acad. Sci. USA 2009, 106, 12986–12991. [Google Scholar] [CrossRef]
- Coulibaly, F.; Chevalier, C.; Gutsche, I.; Pous, J.; Navaza, J.; Bressanelli, S.; Delmas, B.; Rey, F.A. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 2005, 120, 761–772. [Google Scholar] [CrossRef]
- Schofield, D.J.; Glamann, J.; Emerson, S.U.; Purcell, R.H. Identification by phage display and characterization of two neutralizing chimpanzee monoclonal antibodies to the hepatitis E virus capsid protein. J. Virol. 2000, 74, 5548–5555. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, B.; Sun, Y.; Li, H.; Du, T.; Nan, Y.; Hiscox, J.A.; Zhou, E.M.; Zhao, Q. Characterization of Three Novel Linear Neutralizing B-Cell Epitopes in the Capsid Protein of Swine Hepatitis E Virus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Zhao, M.; Li, X.J.; Tang, Z.M.; Yang, F.; Wang, S.L.; Cai, W.; Zhang, K.; Xia, N.S.; Zheng, Z.Z. A Comprehensive Study of Neutralizing Antigenic Sites on the Hepatitis E Virus (HEV) Capsid by Constructing, Clustering, and Characterizing a Tool Box. J. Biol. Chem. 2015, 290, 19910–19922. [Google Scholar] [CrossRef]
- Tang, Z.M.; Tang, M.; Zhao, M.; Wen, G.P.; Yang, F.; Cai, W.; Wang, S.L.; Zheng, Z.Z.; Xia, N.S. A novel linear neutralizing epitope of hepatitis E virus. Vaccine 2015, 33, 3504–3511. [Google Scholar] [CrossRef]
- Xiaofang, L.; Zafrullah, M.; Ahmad, F.; Jameel, S. A C-Terminal Hydrophobic Region is Required for Homo-Oligomerization of the Hepatitis E Virus Capsid (ORF2) Protein. J. Biomed. Biotechnol. 2001, 1, 122–128. [Google Scholar] [CrossRef]
- Li, S.; Tang, X.; Seetharaman, J.; Yang, C.; Gu, Y.; Zhang, J.; Du, H.; Shih, J.W.; Hew, C.L.; Sivaraman, J.; et al. Dimerization of hepatitis E virus capsid protein E2s domain is essential for virus-host interaction. PLoS Pathog. 2009, 5, e1000537. [Google Scholar] [CrossRef]
- Aggarwal, R.; Shukla, R.; Jameel, S.; Agrawal, S.; Puri, P.; Gupta, V.K.; Patil, A.P.; Naik, S. T-cell epitope mapping of ORF2 and ORF3 proteins of human hepatitis E virus. J. Viral Hepat. 2007, 14, 283–292. [Google Scholar] [CrossRef]
- Chen, H.Y.; Lu, Y.; Howard, T.; Anderson, D.; Fong, P.Y.; Hu, W.P.; Chia, C.P.; Guan, M. Comparison of a new immunochromatographic test to enzyme-linked immunosorbent assay for rapid detection of immunoglobulin m antibodies to hepatitis e virus in human sera. Clin. Diagn. Lab. Immunol. 2005, 12, 593–598. [Google Scholar] [CrossRef]
- Hu, W.P.; Lu, Y.; Precioso, N.A.; Chen, H.Y.; Howard, T.; Anderson, D.; Guan, M. Double-antigen enzyme-linked immunosorbent assay for detection of hepatitis E virus-specific antibodies in human or swine sera. Clin. Vaccine Immunol. 2008, 15, 1151–1157. [Google Scholar] [CrossRef][Green Version]
- Mazalovska, M.; Varadinov, N.; Koynarski, T.; Minkov, I.; Teoharov, P.; Lomonossoff, G.P.; Zahmanova, G. Detection of Serum Antibodies to Hepatitis E Virus Based on HEV Genotype 3 ORF2 Capsid Protein Expressed in Nicotiana benthamiana. Ann. Lab. Med. 2017, 37, 313–319. [Google Scholar] [CrossRef][Green Version]
- Taherkhani, R.; Makvandi, M.; Farshadpour, F. Development of enzyme-linked immunosorbent assays using 2 truncated ORF2 proteins for detection of IgG antibodies against hepatitis E virus. Ann. Lab. Med. 2014, 34, 118–126. [Google Scholar] [CrossRef]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223. [Google Scholar] [CrossRef]
- Ankavay, M.; Montpellier, C.; Sayed, I.M.; Saliou, J.M.; Wychowski, C.; Saas, L.; Duvet, S.; Aliouat-Denis, C.M.; Farhat, R.; de Masson d’Autume, V.; et al. New insights into the ORF2 capsid protein, a key player of the hepatitis E virus lifecycle. Sci. Rep. 2019, 9, 6243. [Google Scholar] [CrossRef]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef] [PubMed]
- Osterman, A.; Vizoso Pinto, M.G.; Haase, R.; Nitschko, H.; Jager, S.; Sander, M.; Motz, M.; Mohn, U.; Baiker, A. Systematic screening for novel, serologically reactive Hepatitis E Virus epitopes. Virol. J. 2012, 9, 28. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, S.; Nan, Y.; Ma, Z.; Yang, L.; Zhang, Y. A Linear Surface Epitope in a Proline-Rich Region of ORF3 Product of Genotype 1 Hepatitis E Virus. Viruses 2016, 8, 227. [Google Scholar] [CrossRef]
- Okamoto, H. Efficient cell culture systems for hepatitis E virus strains in feces and circulating blood. Rev. Med. Virol. 2011, 21, 18–31. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, F.; Zhang, L.; Harrison, T.J.; Huang, W.; Zhao, C.; Kong, W.; Jiang, C.; Wang, Y. Hepatitis E Virus Produced from Cell Culture Has a Lipid Envelope. PLoS ONE 2015, 10, e0132503. [Google Scholar] [CrossRef] [PubMed]
- Zafrullah, M.; Ozdener, M.H.; Panda, S.K.; Jameel, S. The ORF3 protein of hepatitis E virus is a phosphoprotein that associates with the cytoskeleton. J. Virol. 1997, 71, 9045–9053. [Google Scholar] [CrossRef]
- Tyagi, S.; Korkaya, H.; Zafrullah, M.; Jameel, S.; Lal, S.K. The phosphorylated form of the ORF3 protein of hepatitis E virus interacts with its non-glycosylated form of the major capsid protein, ORF2. J. Biol. Chem. 2002, 277, 22759–22767. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Nguyen, H.; Yu, C.; Elkins, W.R.; St Claire, M.; Purcell, R.H.; Emerson, S.U. The open reading frame 3 gene of hepatitis E virus contains a cis-reactive element and encodes a protein required for infection of macaques. J. Virol. 2005, 79, 6680–6689. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Opriessnig, T.; Halbur, P.G.; Meng, X.J. Initiation at the third in-frame AUG codon of open reading frame 3 of the hepatitis E virus is essential for viral infectivity in vivo. J. Virol. 2007, 81, 3018–3026. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Pollan, A.; Abrami, L.; Oechslin, N.; Mauron, J.; Matter, M.; Oppliger, J.; Szkolnicka, D.; Dao Thi, V.L.; van der Goot, F.G.; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog. 2018, 14, e1007471. [Google Scholar] [CrossRef]
- Kannan, H.; Fan, S.; Patel, D.; Bossis, I.; Zhang, Y.J. The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J. Virol. 2009, 83, 6375–6382. [Google Scholar] [CrossRef]
- Emerson, S.U.; Nguyen, H.T.; Torian, U.; Burke, D.; Engle, R.; Purcell, R.H. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J. Virol. 2010, 84, 9059–9069. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Jirintai; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 2011, 92, 269–278. [Google Scholar] [CrossRef]
- Kenney, S.P.; Wentworth, J.L.; Heffron, C.L.; Meng, X.J. Replacement of the hepatitis E virus ORF3 protein PxxP motif with heterologous late domain motifs affects virus release via interaction with TSG101. Virology 2015, 486, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef]
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705. [Google Scholar] [CrossRef]
- Lei, Q.; Li, L.; Zhang, S.; Li, T.; Zhang, X.; Ding, X.; Qin, B. HEV ORF3 downregulates TLR7 to inhibit the generation of type I interferon via impairment of multiple signaling pathways. Sci. Rep. 2018, 8, 8585. [Google Scholar] [CrossRef]
- Farhat, R.; Ankavay, M.; Lebsir, N.; Gouttenoire, J.; Jackson, C.L.; Wychowski, C.; Moradpour, D.; Dubuisson, J.; Rouille, Y.; Cocquerel, L. Identification of GBF1 as a cellular factor required for hepatitis E virus RNA replication. Cell Microbiol. 2018, 20. [Google Scholar] [CrossRef]
- Yin, X.; Feng, Z. Hepatitis E Virus Entry. Viruses 2019, 11, 883. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Meng, X.J. Hepatitis E virus replication requires an active ubiquitin-proteasome system. J. Virol. 2012, 86, 5948–5952. [Google Scholar] [CrossRef]
- Surjit, M.; Jameel, S.; Lal, S.K. The ORF2 protein of hepatitis E virus binds the 5′ region of viral RNA. J. Virol. 2004, 78, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef]
- Ju, X.; Ding, Q. Hepatitis E Virus Assembly and Release. Viruses 2019, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- van de Garde, M.D.; Pas, S.D.; van der Net, G.; de Man, R.A.; Osterhaus, A.D.; Haagmans, B.L.; Boonstra, A.; Vanwolleghem, T. Hepatitis E Virus (HEV) Genotype 3 Infection of Human Liver Chimeric Mice as a Model for Chronic HEV Infection. J. Virol. 2016, 90, 4394–4401. [Google Scholar] [CrossRef] [PubMed]
- Marion, O.; Capelli, N.; Lhomme, S.; Dubois, M.; Pucelle, M.; Abravanel, F.; Kamar, N.; Izopet, J. Hepatitis E virus genotype 3 and capsid protein in the blood and urine of immunocompromised patients. J. Infect. 2019, 78, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Himmelsbach, K.; Bender, D.; Hildt, E. Life cycle and morphogenesis of the hepatitis E virus. Emerg. Microbes Infect. 2018, 7, 196. [Google Scholar] [CrossRef]
- Krain, L.J.; Nelson, K.E.; Labrique, A.B. Host immune status and response to hepatitis E virus infection. Clin. Microbiol. Rev. 2014, 27, 139–165. [Google Scholar] [CrossRef]
- Favorov, M.O.; Khudyakov, Y.E.; Mast, E.E.; Yashina, T.L.; Shapiro, C.N.; Khudyakova, N.S.; Jue, D.L.; Onischenko, G.G.; Margolis, H.S.; Fields, H.A. IgM and IgG antibodies to hepatitis E virus (HEV) detected by an enzyme immunoassay based on an HEV-specific artificial recombinant mosaic protein. J. Med. Virol. 1996, 50, 50–58. [Google Scholar] [CrossRef]
- Frias, M.; Lopez-Lopez, P.; Zafra, I.; Caballero-Gomez, J.; Machuca, I.; Camacho, A.; Risalde, M.A.; Rivero-Juarez, A.; Rivero, A. Development and clinical validation of a pangenotypic PCR-based assay for the detection and quantification of hepatitis E virus (Orthopevirus A genus). J. Clin. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.B.; Gupta, P.; Durgapal, H.; Rath, S.; Gupta, S.D.; Acharya, S.K.; Panda, S.K. Study of cellular immune response against Hepatitis E virus (HEV). J. Viral Hepat. 2011, 18, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.; Goel, A.; Rawat, A.; Naik, S.; Aggarwal, R. Histological and immunohistochemical features in fatal acute fulminant hepatitis E. Indian J. Pathol. Microbiol. 2012, 55, 22–27. [Google Scholar] [CrossRef]
- Suneetha, P.V.; Pischke, S.; Schlaphoff, V.; Grabowski, J.; Fytili, P.; Gronert, A.; Bremer, B.; Markova, A.; Jaroszewicz, J.; Bara, C.; et al. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology 2012, 55, 695–708. [Google Scholar] [CrossRef]
- Turvey, S.E.; Broide, D.H. Innate immunity. J. Allergy Clin. Immunol. 2010, 125, S24–S32. [Google Scholar] [CrossRef]
- Brown, A.; Halliday, J.S.; Swadling, L.; Madden, R.G.; Bendall, R.; Hunter, J.G.; Maggs, J.; Simmonds, P.; Smith, D.B.; Vine, L.; et al. Characterization of the Specificity, Functionality, and Durability of Host T-Cell Responses Against the Full-Length Hepatitis E Virus. Hepatology 2016, 64, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Soon, C.F.; Behrendt, P.; Todt, D.; Manns, M.P.; Wedemeyer, H.; Sallberg Chen, M.; Cornberg, M. Defining virus-specific CD8+ TCR repertoires for therapeutic regeneration of T cells against chronic hepatitis E. J. Hepatol. 2019, 71, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Francois, C.; Anggakusuma; Behrendt, P.; Engelmann, M.; Knegendorf, L.; Vieyres, G.; Wedemeyer, H.; Hartmann, R.; Pietschmann, T.; et al. Antiviral Activities of Different Interferon Types and Subtypes against Hepatitis E Virus Replication. Antimicrob. Agents Chemother. 2016, 60, 2132–2139. [Google Scholar] [CrossRef]
- Hardy, M.P.; Owczarek, C.M.; Jermiin, L.S.; Ejdeback, M.; Hertzog, P.J. Characterization of the type I interferon locus and identification of novel genes. Genomics 2004, 84, 331–345. [Google Scholar] [CrossRef]
- Xi, Y.; Day, S.L.; Jackson, R.J.; Ranasinghe, C. Role of novel type I interferon epsilon in viral infection and mucosal immunity. Mucosal Immunol. 2012, 5, 610–622. [Google Scholar] [CrossRef]
- Olagnier, D.; Hiscott, J. Type I and type III interferon-induced immune response: It’s a matter of kinetics and magnitude. Hepatology 2014, 59, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Asselin-Paturel, C.; Brizard, G.; Chemin, K.; Boonstra, A.; O’Garra, A.; Vicari, A.; Trinchieri, G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J. Exp. Med. 2005, 201, 1157–1167. [Google Scholar] [CrossRef]
- Nan, Y.; Nan, G.; Zhang, Y.J. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 2014, 6, 4999–5027. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbiol. 2010, 13, 503–507. [Google Scholar] [CrossRef]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2013, 2, e00785. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Zhang, Y.J. Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Sakai, K.; Matsumoto, M.; Seya, T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 2010, 40, 940–948. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ha, Y.; Chae, C. Expression of interferon-alpha and Mx protein in the livers of pigs experimentally infected with swine hepatitis E virus. J. Comp. Pathol. 2010, 142, 187–192. [Google Scholar] [CrossRef]
- Devhare, P.B.; Desai, S.; Lole, K.S. Innate immune responses in human hepatocyte-derived cell lines alter genotype 1 hepatitis E virus replication efficiencies. Sci. Rep. 2016, 6, 26827. [Google Scholar] [CrossRef]
- Lin, S.; Yang, Y.; Nan, Y.; Ma, Z.; Yang, L.; Zhang, Y.J. The Capsid Protein of Hepatitis E Virus Inhibits Interferon Induction via Its N-terminal Arginine-Rich Motif. Viruses 2019, 11, 1050. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chang, P.; He, J.; Coyaud, E.; Pierce, B.; Zhang, Y. RNA Helicase DDX3 Interacts with the Capsid Protein of Hepatitis E Virus and Plays an Indispensable Role in the Viral Replication. Preprints 2020. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Srivastava, R.; Aggarwal, R.; Jameel, S.; Puri, P.; Gupta, V.K.; Ramesh, V.S.; Bhatia, S.; Naik, S. Cellular immune responses in acute hepatitis E virus infection to the viral open reading frame 2 protein. Viral Immunol. 2007, 20, 56–65. [Google Scholar] [CrossRef]
- Kumar, A.; Devi, S.G.; Kar, P.; Agarwal, S.; Husain, S.A.; Gupta, R.K.; Sharma, S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014, 65, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef]
- Bolen, C.R.; Ding, S.; Robek, M.D.; Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Koltsida, O.; Hausding, M.; Stavropoulos, A.; Koch, S.; Tzelepis, G.; Ubel, C.; Kotenko, S.V.; Sideras, P.; Lehr, H.A.; Tepe, M.; et al. IL-28A (IFN-lambda2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 2011, 3, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Rogee, S.; Le Gall, M.; Chafey, P.; Bouquet, J.; Cordonnier, N.; Frederici, C.; Pavio, N. Quantitative proteomics identifies host factors modulated during acute hepatitis E virus infection in the swine model. J. Virol. 2015, 89, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Pu, X.; Wang, Y.; Xu, N. Impaired plasma lipid profiles in acute hepatitis. Lipids Health Dis. 2010, 9, 5. [Google Scholar] [CrossRef]
- Kanade, G.D.; Pingale, K.D.; Karpe, Y.A. Protein Interactions Network of Hepatitis E Virus RNA and Polymerase with Host Proteins. Front. Microbiol. 2019, 10, 2501. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, W.; Yang, J.; Wen, Z.; Geng, Y.; Zhao, C.; Zhang, H.; Wang, Y. Systematic identification of hepatitis E virus ORF2 interactome reveals that TMEM134 engages in ORF2-mediated NF-kappaB pathway. Virus Res. 2017, 228, 102–108. [Google Scholar] [CrossRef]
- Subramani, C.; Nair, V.P.; Anang, S.; Mandal, S.D.; Pareek, M.; Kaushik, N.; Srivastava, A.; Saha, S.; Shalimar; Nayak, B.; et al. Host-Virus Protein Interaction Network Reveals the Involvement of Multiple Host Processes in the Life Cycle of Hepatitis E Virus. MSystems 2018, 3. [Google Scholar] [CrossRef]
- Alonzi, T.; Maritano, D.; Gorgoni, B.; Rizzuto, G.; Libert, C.; Poli, V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell Biol. 2001, 21, 1621–1632. [Google Scholar] [CrossRef]
- Chandra, V.; Kar-Roy, A.; Kumari, S.; Mayor, S.; Jameel, S. The hepatitis E virus ORF3 protein modulates epidermal growth factor receptor trafficking, STAT3 translocation, and the acute-phase response. J. Virol. 2008, 82, 7100–7110. [Google Scholar] [CrossRef]
- Chandra, V.; Kalia, M.; Hajela, K.; Jameel, S. The ORF3 protein of hepatitis E virus delays degradation of activated growth factor receptors by interacting with CIN85 and blocking formation of the Cbl-CIN85 complex. J. Virol. 2010, 84, 3857–3867. [Google Scholar] [CrossRef]
- Tyagi, S.; Surjit, M.; Roy, A.K.; Jameel, S.; Lal, S.K. The ORF3 protein of hepatitis E virus interacts with liver-specific alpha1-microglobulin and its precursor alpha1-microglobulin/bikunin precursor (AMBP) and expedites their export from the hepatocyte. J. Biol. Chem. 2004, 279, 29308–29319. [Google Scholar] [CrossRef]
- Spina, A.; Lenglet, A.; Beversluis, D.; de Jong, M.; Vernier, L.; Spencer, C.; Andayi, F.; Kamau, C.; Vollmer, S.; Hogema, B.; et al. A large outbreak of Hepatitis E virus genotype 1 infection in an urban setting in Chad likely linked to household level transmission factors, 2016-2017. PLoS ONE 2017, 12, e0188240. [Google Scholar] [CrossRef]
- Purcell, R.H.; Emerson, S.U. Hepatitis E: An emerging awareness of an old disease. J. Hepatol. 2008, 48, 494–503. [Google Scholar] [CrossRef]
- Azman, A.S.; Ciglenecki, I.; Oeser, C.; Said, B.; Tedder, R.S.; Ijaz, S. The incubation period of hepatitis E genotype 1: Insights from pooled analyses of travellers. Epidemiol. Infect. 2018, 146, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Kini, D.; Sofat, S.; Naik, S.R.; Krawczynski, K. Duration of viraemia and faecal viral excretion in acute hepatitis E. Lancet 2000, 356, 1081–1082. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, X.; Jiang, H.; Yan, Q.; Ai, X.; Wang, Y.; Cai, J.; Jiang, L.; Wu, T.; Wang, Z.; et al. Profile of acute infectious markers in sporadic hepatitis E. PLoS ONE 2010, 5, e13560. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.K.; Ansari, I.H.; Acharya, S.K.; Jameel, S.; Panda, S.K. Protracted viremia during acute sporadic hepatitis E virus infection. Gastroenterology 1995, 108, 225–230. [Google Scholar] [CrossRef]
- Chandra, N.S.; Sharma, A.; Malhotra, B.; Rai, R.R. Dynamics of HEV viremia, fecal shedding and its relationship with transaminases and antibody response in patients with sporadic acute hepatitis E. Virol. J. 2010, 7, 213. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, F.M.; Di Bartolo, I.; Ponterio, E.; Angeloni, G.; Trevisani, M.; Ostanello, F. Zoonotic transmission of hepatitis E virus in industrialized countries. New Microbiol. 2013, 36, 331–344. [Google Scholar] [PubMed]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Peron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef]
- Zhou, X.; de Man, R.A.; de Knegt, R.J.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Epidemiology and management of chronic hepatitis E infection in solid organ transplantation: A comprehensive literature review. Rev. Med. Virol. 2013, 23, 295–304. [Google Scholar] [CrossRef]
- Ankcorn, M.J.; Ijaz, S.; Poh, J.; Elsharkawy, A.M.; Smit, E.; Cramb, R.; Ravi, S.; Martin, K.; Tedder, R.; Neuberger, J. Toward Systematic Screening for Persistent Hepatitis E Virus Infections in Transplant Patients. Transplantation 2018, 102, 1139–1147. [Google Scholar] [CrossRef]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus quasispecies and the outcome of acute hepatitis E in solid-organ transplant patients. J. Virol. 2012, 86, 10006–10014. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H. Current status of hepatitis e virus infection in Korea. Gut Liver 2011, 5, 427–431. [Google Scholar] [CrossRef][Green Version]
- Colson, P.; Romanet, P.; Moal, V.; Borentain, P.; Purgus, R.; Benezech, A.; Motte, A.; Gerolami, R. Autochthonous infections with hepatitis E virus genotype 4, France. Emerg. Infect. Dis. 2012, 18, 1361–1364. [Google Scholar] [CrossRef]
- Narayanan, S.; Abutaleb, A.; Sherman, K.E.; Kottilil, S. Clinical features and determinants of chronicity in hepatitis E virus infection. J. Viral Hepat. 2019, 26, 414–421. [Google Scholar] [CrossRef]
- Anty, R.; Ollier, L.; Peron, J.M.; Nicand, E.; Cannavo, I.; Bongain, A.; Giordanengo, V.; Tran, A. First case report of an acute genotype 3 hepatitis E infected pregnant woman living in South-Eastern France. J. Clin. Virol. 2012, 54, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Lachish, T.; Erez, O.; Daudi, N.; Shouval, D.; Schwartz, E. Acute hepatitis E virus in pregnant women in Israel and in other industrialized countries. J. Clin. Virol. 2015, 73, 20–24. [Google Scholar] [CrossRef]
- Deroux, A.; Brion, J.P.; Hyerle, L.; Belbezier, A.; Vaillant, M.; Mosnier, E.; Larrat, S.; Morand, P.; Pavese, P. Association between hepatitis E and neurological disorders: Two case studies and literature review. J. Clin. Virol. 2014, 60, 60–62. [Google Scholar] [CrossRef]
- Wu, X.; Liu, K.; Zhang, H.L. Guillain-Barre syndrome and encephalitis/encephalopathy associated with acute severe hepatitis E infection. Neurol. Sci. 2015, 36, 165–166. [Google Scholar] [CrossRef]
- Jha, A.K.; Nijhawan, S.; Nepalia, S.; Suchismita, A. Association of Bell’s Palsy with Hepatitis E Virus Infection: A Rare Entity. J. Clin. Exp. Hepatol. 2012, 2, 88–90. [Google Scholar] [CrossRef]
- Fong, F.; Illahi, M. Neuralgic amyotrophy associated with hepatitis E virus. Clin. Neurol. Neurosurg. 2009, 111, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Perrin, H.B.; Cintas, P.; Abravanel, F.; Gerolami, R.; d’Alteroche, L.; Raynal, J.-N.; Alric, L.; Dupuis, E.; Prudhomme, L.; Vaucher, E.; et al. Neurologic Disorders in Immunocompetent Patients with Autochthonous Acute Hepatitis E. Emerg. Infect. Dis. 2015, 21, 1928–1934. [Google Scholar] [CrossRef]
- Shi, R.; Soomro, M.H.; She, R.; Yang, Y.; Wang, T.; Wu, Q.; Li, H.; Hao, W. Evidence of Hepatitis E virus breaking through the blood-brain barrier and replicating in the central nervous system. J. Viral Hepat. 2016, 23, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Pique, J.; Couturier, E.; Nicot, F.; Dimeglio, C.; Lhomme, S.; Chiabrando, J.; Saune, K.; Peron, J.M.; Kamar, N.; et al. Acute hepatitis E in French patients and neurological manifestations. J. Infect. 2018, 77, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Ripellino, P.; Pasi, E.; Melli, G.; Staedler, C.; Fraga, M.; Moradpour, D.; Sahli, R.; Aubert, V.; Martinetti, G.; Bihl, F.; et al. Neurologic complications of acute hepatitis E virus infection. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e643. [Google Scholar] [CrossRef]
- Bura, M.; Michalak, M.; Chojnicki, M.; Czajka, A.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Seroprevalence of anti-HEV IgG in 182 Polish patients. Postepy Hig. Med. Dosw. 2015, 69, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Bazerbachi, F.; Haffar, S.; Garg, S.K.; Lake, J.R. Extra-hepatic manifestations associated with hepatitis E virus infection: A comprehensive review of the literature. Gastroenterol. Rep. 2016, 4, 1–15. [Google Scholar] [CrossRef]
- Cheng, S.H.; Mai, L.; Zhu, F.Q.; Pan, X.F.; Sun, H.X.; Cao, H.; Shu, X.; Ke, W.M.; Li, G.; Xu, Q.H. Influence of chronic HBV infection on superimposed acute hepatitis E. World J. Gastroenterol. 2013, 19, 5904–5909. [Google Scholar] [CrossRef]
- Divizia, M.; Gabrieli, R.; Stefanoni, M.L.; Renganathan, E.; El Ghazzawi, E.; Kader, O.A.; Gamil, F.; El Sawaf, G.; El Sherbini, E.; Saleh, E.; et al. HAV and HEV infection in hospitalised hepatitis patients in Alexandria, Egypt. Eur. J. Epidemiol. 1999, 15, 603–609. [Google Scholar] [CrossRef]
- Asaei, S.; Ziyaeyan, M.; Moeini, M.; Jamalidoust, M.; Behzadi, M.A. Seroprevalence of Hepatitis A and E Virus Infections among Healthy Population in Shiraz, Southern Iran. Jundishapur J. Microbiol. 2015, 8, e19311. [Google Scholar] [CrossRef]
- Oh, H.W.; Cha, R.R.; Lee, S.S.; Lee, C.M.; Kim, W.S.; Jo, Y.W.; Kim, J.J.; Lee, J.M.; Kim, H.J.; Ha, C.Y.; et al. Comparing the Clinical Features and Outcomes of Acute Hepatitis E Viral Infections with Those of Acute Hepatitis A, B, and C Infections in Korea. Intervirology 2017, 60, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Greco, L.; Uceda Renteria, S.C.; Guarneri, D.; Orlandi, A.; Zoccoli, A.; Benardon, S.; Cusini, M.; Lunghi, G. HEV and HAV seroprevalence in men that have sex with men (MSM): An update from Milan, Italy. J. Med. Virol. 2018, 90, 1323–1327. [Google Scholar] [CrossRef]
- Joon, A.; Rao, P.; Shenoy, S.M.; Baliga, S. Prevalence of Hepatitis A virus (HAV) and Hepatitis E virus (HEV) in the patients presenting with acute viral hepatitis. Indian J. Med. Microbiol. 2015, 33, 102–105. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Z.; Lv, J.; Liu, J.; Yan, B.; Feng, Y.; Li, L.; Zhang, G.; Wang, F.; Xu, A. Comparison of hepatitis E virus seroprevalence between HBsAg-positive population and healthy controls in Shandong province, China. BMC Infect. Dis. 2018, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Bayram, A.; Eksi, F.; Mehli, M.; Sozen, E. Prevalence of hepatitis E virus antibodies in patients with chronic hepatitis B and chronic hepatitis C. Intervirology 2007, 50, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, S.Y.; Zhang, D.D.; Li, X.Y.; Zhang, Y.L.; Li, W.X.; Yan, J.J.; Wang, M.; Xun, J.N.; Lu, C.; et al. Clinical features of acute hepatitis E super-infections on chronic hepatitis B. World J. Gastroenterol. 2016, 22, 10388–10397. [Google Scholar] [CrossRef] [PubMed]
- Hoan, N.X.; Tong, H.V.; Hecht, N.; Sy, B.T.; Marcinek, P.; Meyer, C.G.; Song le, H.; Toan, N.L.; Kurreck, J.; Kremsner, P.G.; et al. Hepatitis E Virus Superinfection and Clinical Progression in Hepatitis B Patients. EBioMedicine 2015, 2, 2080–2086. [Google Scholar] [CrossRef]
- Koning, L.; Charlton, M.R.; Pas, S.D.; Heimbach, J.K.; Osterhaus, A.D.; Watt, K.D.; Janssen, H.L.; de Knegt, R.J.; van der Eijk, A.A. Prevalence and clinical consequences of Hepatitis E in patients who underwent liver transplantation for chronic Hepatitis C in the United States. BMC Infect. Dis. 2015, 15, 371. [Google Scholar] [CrossRef]
- Chen, Y.D.; Liu, M.Y.; Yu, W.L.; Li, J.Q.; Dai, Q.; Zhou, Z.Q.; Tisminetzky, S.G. Mix-infections with different genotypes of HCV and with HCV plus other hepatitis viruses in patients with hepatitis C in China. World J. Gastroenterol. 2003, 9, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Yugo, D.M.; Hauck, R.; Shivaprasad, H.L.; Meng, X.J. Hepatitis Virus Infections in Poultry. Avian Dis. 2016, 60, 576–588. [Google Scholar] [CrossRef]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef]
- Dibba, P.; Cholankeril, R.; Li, A.A.; Patel, M.; Fayek, M.; Dibble, C.; Okpara, N.; Hines, A.; Ahmed, A. Hepatitis C in Pregnancy. Diseases 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T. Hepatitis B in Pregnancy. Clin. Infect. Dis. 2016, 62 (Suppl. 4), S314–S317. [Google Scholar] [CrossRef]
- Barnett, M.A.; Learmonth, R.P.; Pihl, E.; Wood, E.C. T helper lymphocyte depression in early human pregnancy. J. Reprod. Immunol. 1983, 5, 55–57. [Google Scholar] [CrossRef]
- Malinowski, A.; Szpakowski, M.; Tchorzewski, H.; Zeman, K.; Pawlowicz, P.; Wozniak, P. T lymphocyte subpopulations and lymphocyte proliferative activity in normal and pre-eclamptic pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1994, 53, 27–31. [Google Scholar] [CrossRef]
- Degenne, D.; Canepa, S.; Lecomte, C.; Renoux, M.; Bardos, P. Serial study of T-lymphocyte subsets in women during very early pregnancy. J. Clin. Immunol. 1988, 48, 187–191. [Google Scholar] [CrossRef]
- Sykes, L.; MacIntyre, D.A.; Yap, X.J.; Teoh, T.G.; Bennett, P.R. The Th1:th2 dichotomy of pregnancy and preterm labour. Mediat. Inflamm. 2012, 2012, 967629. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, M.G.; Chaouat, G.; Florez, L.; Bensussan, A.; Klatzmann, D. Regulatory T-cells in pregnancy: Historical perspective, state of the art, and burning questions. Front. Immunol. 2014, 5, 389. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Aggarwal, R.; Naik, S.R.; Das, V.; Das, S.; Naik, S. Immunological alterations in pregnant women with acute hepatitis E. J. Gastroenterol. Hepatol. 2005, 20, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Patra, S.; David, P.; Vyas, A.; Khanam, A.; Hissar, S.; Gupta, E.; Kumar, G.; Kottilil, S.; Maiwall, R.; et al. Impaired monocyte-macrophage functions and defective Toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology 2015, 62, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Knegendorf, L.; Drave, S.A.; Dao Thi, V.L.; Debing, Y.; Brown, R.J.P.; Vondran, F.W.R.; Resner, K.; Friesland, M.; Khera, T.; Engelmann, M.; et al. Hepatitis E virus replication and interferon responses in human placental cells. Hepatol. Commun. 2018, 2, 173–187. [Google Scholar] [CrossRef]
- Jilani, N.; Das, B.C.; Husain, S.A.; Baweja, U.K.; Chattopadhya, D.; Gupta, R.K.; Sardana, S.; Kar, P. Hepatitis E virus infection and fulminant hepatic failure during pregnancy. J. Gastroenterol. Hepatol. 2007, 22, 676–682. [Google Scholar] [CrossRef]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef]
- Rijhsinghani, A.G.; Thompson, K.; Bhatia, S.K.; Waldschmidt, T.J. Estrogen blocks early T cell development in the thymus. Am. J. Reprod. Immunol. 1996, 36, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, T.A.; DeMayo, F.; Rich, S.; Conneely, O.M.; O’Malley, B.W. Progesterone receptors in the thymus are required for thymic involution during pregnancy and for normal fertility. Proc. Natl. Acad. Sci. USA 1999, 96, 12021–12026. [Google Scholar] [CrossRef]
- Yang, C.; Yu, W.; Bi, Y.; Long, F.; Li, Y.; Wei, D.; Hao, X.; Situ, J.; Zhao, Y.; Huang, F. Increased oestradiol in hepatitis E virus-infected pregnant women promotes viral replication. J. Viral Hepat. 2018, 25, 742–751. [Google Scholar] [CrossRef]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Bruggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, S.; He, Q.; Liang, Z.; Wang, L.; Wang, Q.; Wang, L. Hepatitis E-related adverse pregnancy outcomes and their prevention by hepatitis E vaccine in a rabbit model. Emerg. Microbes Infect. 2019, 8, 1066–1075. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form a | Start Codon | Genome Association | Glycosylation | Secretion |
|---|---|---|---|---|
| ORF2c | Met15 | Yes | No | No |
| ORF2s | Met1 | No | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; Zhang, Y.-J. Advances in Hepatitis E Virus Biology and Pathogenesis. Viruses 2021, 13, 267. https://doi.org/10.3390/v13020267
Lin S, Zhang Y-J. Advances in Hepatitis E Virus Biology and Pathogenesis. Viruses. 2021; 13(2):267. https://doi.org/10.3390/v13020267
Chicago/Turabian StyleLin, Shaoli, and Yan-Jin Zhang. 2021. "Advances in Hepatitis E Virus Biology and Pathogenesis" Viruses 13, no. 2: 267. https://doi.org/10.3390/v13020267
APA StyleLin, S., & Zhang, Y.-J. (2021). Advances in Hepatitis E Virus Biology and Pathogenesis. Viruses, 13(2), 267. https://doi.org/10.3390/v13020267